
Basics of Nitrosamine Impurities Quality Risk Management

Background Nitrosamine Impurities:
Authorities in the EU discovered a nitrosamine impurity, N-nitrosodimethylamine (NDMA), in valsartan from one manufacturer of active pharmaceutical ingredient (API) in June 2018. N-nitrosodiethylamine (NDEA), another nitrosamine, was later discovered.
The FDA discovered that three popular heartburn medications (ranitidine, also known as Zantac, and nizatidine, also known as Axid) contained unsafe levels of NDMA in September 2019.
The FDA discovered in December 2019 that some Metformin diabetes medications in other nations were reportedly found to contain NDMA.
The FDA then discovered seven nitrosamine impurities that could potentially be found in medication products:
| Sr. No. | Nitrosamine | Chemical name | CSA No. | Chemical Structure |
| 1. | NDMA | N-nitrosodimethylamine | 62-75-9 |  |
| 2. | NDEA | N-nitrosodiethylamine | 55-18-5 |  |
| 3. | NMBA | N-nitroso-N-methyl-4-aminobutanoic acid | 61445-55-4 | 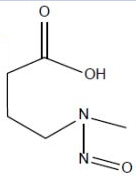 |
| 4. | NMPA | N-nitrosomethylphenylamine | 614-00-6 | 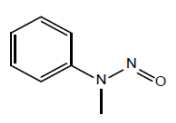 |
| 5. | NIPEA/ EIPNA | N-nitrosoisopropylethyl amine | 16339-04-1 | 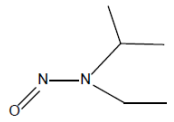 |
| 6. | NDIPA/ DIPNA | N-nitrosodiisopropylamine | 601-77-4 |  |
| 7. | MeNP | 1-Methyl-4-nitrosopiperazine | 16339-07-4 |  |
| 8. | NDBA | N-nitrosodibutylamine | 924-16-3 | 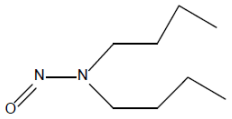 |
According to the ICH M7(R1) guideline, the presence of N-nitrosamines in human pharmaceutical goods must be minimised at or below the acceptable intake (AI) due to their recognised potential carcinogenic effect.
An appropriate control strategy that considers the starting material, intermediates, reagents, catalysts, manufacturing process, excipients, shared equipment, water, disinfectant, solvents, packaging material, and should aim to prevent the formation of and contamination with N-nitrosamines, verification of analytical methods to determine and control the impurities, as well as other factors should be used to limit the possibility of the formation of these impurities in the drug substance and drug product.
Limits for the intake of the impurities based on Acceptable Intake (AI) and Maximum Daily Dose (MDD), shall be calculated and the root cause for the presence of Nitrosamine impurity shall be identified and an appropriate CAPA shall be implemented as needed.
Purpose of Nitrosamine Impurities:
This document’s goal is to review and evaluate the possibility that produced drug substances and drug products in the manufacturing facility may include nitrosamine impurities both in the past and in the future.
Quality risk management approach for nitrosamine impurities:
A class of substances with the chemical structure of a nitroso group bound to an amine are referred to as nitrosamines. A nitrosating reaction between amines (secondary, tertiary, or quaternary amines) and nitrous acid can result in the chemicals (nitrite salts under acidic conditions).
The three stage approach is used for nitrosamine impurity risk evaluation of drug substances and drug products, according to the EMA reference EMA/189634/2019.
Step 1 Risk Evaluation:
All risk variables or factors that can affect the production of nitrosamine impurities are considered in the assessment, including
Presence of Nitrites or other Nitrosating Agents:
When secondary, tertiary, or quaternary amines and nitrite salts are present during an acidic process, nitrosamines may form. In these circumstances, nitrite salts might produce nitrous acid, which could then interact with an amine to create nitrosamine.
Sources of secondary, tertiary quaternary amines:
Amines may come from the API, API degradants, intermediates, or starting materials, reagents, or catalysts in a manufacturing process. To create nitrosamines, these amines can interact with nitrous acid or other nitrosating substances.
Amide Solvents:
Another source of secondary amines is amine solvents, which can degrade under specific reaction circumstances. These secondary amines can create nitrosamines when they interact with nitrous acid.
Contamination in Raw Materials:
- Solvent contamination during shipment
- Starting materials or outsourced intermediates produced in locations where nitrosamine impurities are created in other processes.
- Nitrate or nitrite as impurities in starting materials.
- Impurities of secondary or tertiary amines in various raw materials and fresh solvents.
Recovered Solvents, Catalysts, and Reagents:
Due to the presence of leftover amines, recovered materials such solvents, reagents, and catalysts, including recovery performed by other parties, may represent a danger of nitrosamine contamination. Nitrous acid may be used in solvent recovery processes that result in the synthesis of Nitrosamines.
Lack of Process Optimization and Control:
Inadequately controlled or inappropriate reaction conditions, such as temperature, pH, or the order in which reagents, intermediates, or solvents are added.
Control over Excipients:
A variety of frequently used excipients contain nitrite impurities, which can cause nitrosamine impurities to develop in drug products during the production process and shelf-life storage period.
Cross contamination:
Equipment that is shared among several processes and goods may cause cross contamination when cleaning assurance is inadequate.
Water Quality:
Nitrosamines are produced as a result of the use of chloramine in water. Using oxidants can produce nitrogen dioxide, which reacts with amines to create nitrosamines.
Packaging Materials:
Contamination from blister packaging materials in the lidding foil containing nitrocellulose printing primer may react with amines in printing ink to generate nitrosamines, which would be transferred to the product under certain packaging process conditions
Step 2 Confirmatory Testing for presence of nitrosamines:
- If nitrosamines are found to pose a concern as a consequence of the risk assessment, confirmatory testing should be conducted using sensitive and suitably validated techniques that may be Official analytical method or in-house validated method with adequate LOD and LOQ.
- Identifying the Nitrosamine impurities in the drug substance and drug product based on the risk assessment’s Low/ Medium/ High risk classification.
- Confirmation testing for the presence of nitrosamine impurities in all drug substances and drug products listed as being at high risk.
- Development and deploy an appropriate validated and sensitive method in the facility.
- Investigation of the Nitrosamine impurity, identification of the underlying cause, and implementation of the necessary corrective and preventive measures (CAPA).
- Suitable level of specification for the impurity depending on the drug product’s dosage, duration of use, and claimed allowed daily exposure.
- Defining the control strategy for the presence of Nitrosamine impurity by inclusion in the specifications (Drug Substance or Drug Product) as
- Omission from testing (for LOQ less than 10 % of acceptable limit and root cause is identified and understood).
- Skip Lot Testing (for LOQ more than 10 % but less than 30 % of acceptable limit).
- As regular testing (for LOQ more than 30 % of acceptable limit).
Acceptable Intake (AI) limits for the Nitrosamine Impurities are:
| Nitrosamine | Chemical name | EU – AI Limit (ng/day) | US FDA – AI Limit (ng/day) |
| NDMA | N-nitrosodimethylamine | 96 | 96 |
| NDEA | N-nitrosodiethylamine | 26.5 | 26.5 |
| NMBA | N-nitroso-N-methyl-4-aminobutanoic acid | 96 | 96 |
| NMPA | N-nitrosomethylphenylamine | 34.3 | 26.5 |
| NIPEA/ EIPNA | N-nitrosoisopropylethyl amine | 26.5 | 26.5 |
| NDIPA/ DIPNA | N-nitrosodiisopropylamine | 26.5 | 26.5 |
| MeNP | 1-Methyl-4-nitrosopiperazine | 26.5 | – |
| NDBA | N-nitrosodibutylamine | 26.5 | – |
Note: Limits should be applied to the Maximum Daily Dose
Calculation of acceptable daily limit of Nitrosamine in the API or Formulation:
Acceptable Intake (AI) limit can be calculated in ppm by using the following formula:
Limit = Acceptable Intake (AI)/ Maximum Daily Dose (MDD) (in mg)
API or ingredient having potential for nitrosamine in the formulation having maximum daily dose or intake is 3000 mg
Limit of NDMA can be calculated as follows:
Limit of NDMA in ppm = 96 ng / 3000 mg = 0.032 ppm
If there are 2 or more nitrosamines observed in the API or ingredient having potential for nitrosamine in the formulation, a limit of total nitrosamine also needs to be included. This limit cannot be more than the limit derived using the most stringent nitrosamine.
Eg: If NDMA, NDEA and NDPA are observed in a API or ingredient in the formulation having a MDD of 3000 mg the limit of total impurities will be calculated as:
Total Impurities (in ppm) = AI of most stringent nitrosamine (i.e. 26.5 ng)/ MDD (3000 mg)
Total Impurities (ppm) = 26.5/3000 = 0.009 ppm
Step 3 Changes to the Marketing Authorisation:
Drug substances and drug products should notify the Marketing authorisations or FDA of any proposed changes in the production process to prevent or minimise nitrosamine impurities or change in product specifications.
Mitigation Strategy for nitrosamine impurities:
- Creating systems and settings for reactions that can reliably reduce nitrosamine impurities while being sufficiently managed
- Using bases other than secondary, tertiary, or quaternary amines, amide solvents.
- Implement a control strategy for N-nitrosamines that includes on-going and planned actions to reduce the risk of nitrosamine contamination and production (e.g. change of manufacturing process, change of raw material quality, introduction of appropriate specifications and development of appropriate methods, and measures on the premise and equipment such as cleaning procedures and environmental monitoring)
- Make certain that the active ingredients and excipients in their FPs are produced in accordance with good manufacturing principles.
- Any batch result that exceeds the regulatory expectation threshold needs to be regarded as OOT, and GMP action should be taken.
- Any batch of a drug substance or drug product that is discovered to contain nitrosamine impurities at or above the advised AI should be recalled and prevented from being on the market.
Conclusion of Quality Risk Management of nitrosamine impurities:
The risk assessment of all commercial drug products is established, the root cause is identified, and the necessary mitigating action is taken in accordance with the three step approach of risk assessment for the presence of Nitrosamine impurities in drug substance and drug product manufactured by manufacturer. All future products that manufacturing organization develops, produces, and markets will continue to undergo risk assessments for the existence of Nitrosamine contaminants.
Informative videos on nitrosamine impurity:
1. Nitrosamines risk assessment: why?
3. Nitrosamines risk assessment: step 2
4. How to deal with complex nitrosamines?
References guidelines for nitrosamine impurities from various agencies:
i. European Medicines Agency: Nitrosamine impurities guidance
ii. European Medicines Agency: EMA/189634/2019,EMA/428592/2019,EMA/428592/2019, Rev 03, EMA/369136/2020, EMA/425645/2020, EMA/409815/2020 Rev.11 (29 July 2022)
iii. European Medicines Regulatory: Network approach for the implementation of the CHMP Opinion pursuant to Article 5(3) of Regulation (EC) No 726/2004 for nitrosamine impurities in human medicines, 22 February 2021
iv. European Commission: Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products, 29 July 2022
v. US FDA: Control of Nitrosamine Impurities in Human Drugs, Feb 2021
https://www.fda.gov/media/141720/download
vi. United States Pharmacopoeia (USP): Chapter 〈1469〉 NITROSAMINE IMPURITIES
vii. Pmda: Pharmaceuticals and Medical Devices Agency, Japan
Session 4 Impurities: Mutagenic impurities and more Control of nitrosamine impurities in sartan drugs Office of Generic Drugs, PMDA
https://www.pmda.go.jp/files/000241930.pdf
Click this link to read 350 + Pharmaceutical interview questions and answers