
750+ Pharmaceutical Interview Questions and Answers for the year 2022-23
Technical know-how is a must to clear the pharmaceutical interview. Most of the answers are based on basic knowledge and current pharmaceutical guidelines. Here, we have compiled interview questions and answers, most common during the interview for pharmaceutical jobs for working in an oral solid manufacturing facility, sterile manufacturing facility, quality assurance, microbiology, or validation department.
The questions and answers will help pharma freshers, beginners, and experienced individuals. Freshers and beginners can understand what types of questions could be asked and the answers to those questions. Experienced pharma professionals can refresh their basic knowledge, and the questions and answers will help them while interviewing other pharma professionals during the recruitment process.
- Top 15 interview tips for Pharma Professionals
- Pharmaceutical Interview Questions and Answers on General Topic
- Pharmaceutical Interview Questions and Answers on Production System – Oral Solid Dosage Formulation
- Pharmaceutical Interview Questions and Answers on Production System – Sterile and Injectable Dosage Formulation
- Pharmaceutical Interview Questions and Answers on Validation
- Pharmaceutical Interview Questions and Answers on Microbiology
- Useful Interview Questions and Answers on HPLC and Troubleshooting
- Pharmaceutical interview Questions and Answers for quality control Laboratory

Mostly asked 10+ General Pharmaceutical Interview Questions and Answers for 2021-2022
Most commonly asked general questions and answers for the freshers in the pharmaceutical industry. Even though freshers have good knowledge about subjects, following are few general questions which interviewer will expect that candidate must have to know about as it is very basic knowledge and connecting bridge between student life and industry life.
1. What is the Good Manufacturing Practice (GMP) or Current Good Manufacturing Practice (CGMP)? Provide reference to GMP regulations of different countries.
Quality of pharmaceuticals is important for the patient’s safety and are very carefully regulated by respective country regulators.
GMP or CGMP refers to the Current Good Manufacturing Practice regulations enforced by the respective country regulations.
GMP provides systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the GMP regulations assures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations.
The US FDA uses terminology CGMP. According to the U.S. FDA, “‘C’ in CGMP stands for “current,” requiring companies to use technologies and systems that are up-to-date in order to comply with the regulations. Systems and equipment that may have been “top-of-the-line” to prevent contamination, mix-ups, and errors 10 or 20 years ago may be less than adequate by today’s standards.”
GMP regulations governed by different countries and its reference guidance are detailed as follows. Following table provides reference for most common regulatory bodies. This will provide understanding on how GMP regulations are forced by different regulatory bodies.
| Country | Regulatory Body | Reference regulations under respective country’s law |
| United States | U.S. FDA | Code of Federal Regulations (CFR): 21 CFR Part 210. Current Good Manufacturing Practice in Manufacturing Processing, packing, or Holding of Drugs. 21 CFR Part 211. Current Good Manufacturing Practice for Finished Pharmaceuticals. 21 CFR Part 212. Current Good Manufacturing Practice for Positron Emission Tomography Drugs. 21 CFR Part 600. Biological Products: General. 21 CFR Part 314. For FDA approval to market a new drug. |
| European Union (EU) Countries | European Commission – Health and Food Safety | The entire body of EU medicines legislation (EudraLex) is compiled in “The rules governing medicinal products in the European Union”. Pharmaceutical sector is compiled in Volume 1 and Volume 5 of the publication “The rules governing medicinal products in the European Union”: Volume 1 – EU pharmaceutical legislation for medicinal products for human use Volume 5 – EU pharmaceutical legislation for medicinal products for veterinary use |
| Countries following WHO guidance | World Health Organization (WHO) | WHO good manufacturing practices for pharmaceutical products: main principles, Annex 2, WHO Technical Report Series 986, 2014 |
| India | Ministry of health and family welfare | The drugs and cosmetics act, 1940 and The drugs and cosmetics rules, 1945 Schedule M: Good Manufacturing Practices and requirements of premises, plant and equipment for pharmaceutical products |
| Canada | Health Canada | Good Manufacturing Practices Guidelines by Health Products and Food Branch Inspectorate Good Manufacturing Practices (GMP) refer to Division 2, Part C of the Food and Drug Regulations. The guidelines apply to pharmaceutical, radiopharmaceutical, biological, and veterinary drugs and were developed by Health Canada in consultation with their stakeholders. Division 1A, Part C of the Food and Drug Regulations defines activities for which GMP compliance is to be demonstrated prior to the issuance of an establishment license. Guidance based on PIC/S |
| Japan | Pharmaceuticals and Medical Devices Agency (PMDA) | Pharmaceuticals and Medical Devices Agency (PMDA) was established and came into service on April 1, 2004, under the Law for the Pharmaceuticals and Medical Devices Agency, as a consolidation of the services of the Pharmaceuticals and Medical Devices Evaluation Center of the National Institute of Health Sciences (PMDEC), the Organization for Pharmaceutical Safety and Research (OPSR/KIKO), and part of the Japan Association for the Advancement of Medical Equipment (JAAME). GMP Ministerial Ordinance (Ministerial Ordinance on Standards for Manufacturing Control and Quality Control for Drugs and Quasi-drugs) No. 179, 2004 |
| Australia | Therapeutic Goods Administration | AUSTRALIAN CODE OF GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS Therapeutic Goods Act 1989. This upholds the main objective of the Act, which is to ensure the safety, quality, efficacy and timely supply of therapeutic goods for Australian consumers. Guidance based on PIC/S |
| New Zealand | Medsafe | New Zealand Code of Good Manufacturing Practice for Manufacture and Distribution of Therapeutic Goods Part 1: Manufacture of Pharmaceutical Products (2009) Guidance based on PIC/S |
2. What is Standard Operating Procedure (SOP)?
SOPs are documented Standards Operating Procedure, authorized by the Quality Unit or Quality Assurance department having sets of written instructions to be followed by employees on a day to day basis to carry out operations in a consistent manner to achieve predetermined specification and a quality end-result.
Examples of Standard Operating Procedures are as follows:
– SOP for entry and exit in the manufacturing facility
– SOP for operation and cleaning of compression machine
– SOP for preparation of SOP
– SOP for pest control
– SOP for material receipt
– SOP for preventive maintenance program
– SOP for operation of High Performance Liquid Chromatography (HPLC) instrument
3. What is the typical content of Standard Operating Procedure (SOP)?
Objective or Purpose
Scope
Responsibility
Accountability
Definitions
Abbreviations
Reference
Procedure
List of Annexes
Format for recording Revision history
4. What is Master Formula Record, Master Formula, Manufacturing Formula, and Master Production and Control Record?
Master Formula Record, Master Formula, Manufacturing Formula, and Master Production and Control Record mean the same thing. This is an approved master document that describes the full manufacturing process of the drug product. [Reference: 1]
5. What is drug substance?
Drug substance is an active ingredient that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease or to affect the structure or any function of the human body, but does not include intermediates used in the synthesis of such ingredient. [Reference: 2]
6. What is drug product?
Drug product means a finished dosage form, for example, tablet, capsule, solution, etc., that contains an active drug ingredient generally, but not necessarily, in association with inactive ingredients. The term also includes a finished dosage form that does not contain an active ingredient but is intended to be used as a placebo. [Reference:3]
7. What is Active ingredient?
Active ingredient means any component that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease, or to affect the structure or any function of the body of man or other animals. The term includes those components that may undergo chemical change in the manufacture of the drug product and be present in the drug product in a modified form intended to furnish the specified activity or effect. [Reference:3]
8. Fiber means?
Fiber means any particulate contaminant with a length at least three times greater than its width. [Reference:3]
9. What is inactive ingredient?
Inactive ingredient means any component other than an active ingredient. [Reference:3]
10. What is Gang-printing?
Gang-printed labeling means labeling derived from a sheet of material on which more than one item of labeling is printed. [Reference:3]
11. What is full form of ICH?
Full form of ICH is International Council for Harmonisation (ICH), formerly known as the International Conference on Harmonisation (ICH).
12. ICH Guidelines are divided into how many categories? What are those?
The ICH topics are divided into the four categories below.
Quality Guidelines
Safety Guidelines
Efficacy Guidelines
Multidisciplinary Guidelines
13. How many main topic quality guidelines are published by ICH ?
Q1A – Q1F Stability
Q1A(R2) Stability Testing of New Drug Substances and Products
Q1B Stability Testing : Photostability Testing of New Drug Substances and Products
Q1C Stability Testing for New Dosage Forms
Q1D Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products
Q1E Evaluation of Stability Data
Q1F Stability Data Package for Registration Applications in Climatic Zones III and IV
Q2 Analytical Validation
Q2(R1) Validation of Analytical Procedures: Text and Methodology
Q2(R2)/Q14 EWG Analytical Procedure Development and Revision of Q2 (R1) Analytical Validation
Q3A – Q3E Impurities
Q3A(R2) Impurities in New Drug Substances
Q3B(R2) Impurities in New Drug Products
Q3C(R8) Guideline for Residual Solvents
Q3C(R9) Maintenance EWG Maintenance of the Guideline for Residual Solvents
Q3D(R1) Guideline for Elemental Impurities
Q3D(R2) Maintenance EWG Revision of Q3D(R1) for cutaneous and transdermal products
Q3D training Implementation of Guideline for Elemental Impurities
Q3E EWG Impurity: Assessment and Control of Extractables and Leachables for Pharmaceuticals and Biologics
Q4A – Q4B Pharmacopoeias
Q4A Pharmacopoeial Harmonisation
Q4B Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions
Q4B Annex 1(R1) Residue on Ignition/Sulphated Ash General Chapter
Q4B Annex 2(R1) Test for Extractable Volume of Parenteral Preparations General Chapter
Q4B Annex 3(R1) Test for Particulate Contamination: Sub-Visible Particles General Chapter
Q4B Annex 4A(R1) Microbiological Examination of Non-Sterile Products: Microbial Enumeration Tests General Chapter
Q4B Annex 4B(R1) Microbiological Examination of Non-Sterile Products: Tests for Specified Micro-Organisms General Chapter
4C(R1) Microbiological Examination of Non-Sterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use General Chapter
Q4B Annex 5(R1) Disintegration Test General Chapter
Q4B Annex 6 Uniformity of Dosage Units General Chapter
Q4B Annex 7(R2) Dissolution Test General Chapter
Q4B Annex 8(R1) Sterility Test General Chapter
Q4B Annex 9(R1) Tablet Friability General Chapter
Q4B Annex 10(R1) Polyacrylamide Gel Electrophoresis General Chapter
Q4B Annex 11 Capillary Electrophoresis General Chapter
Q4B Annex 12 Analytical Sieving General Chapter
Q4B Annex 13 Bulk Density and Tapped Density of Powders General Chapter
Q4B Annex 14 Bacterial Endotoxins Test General Chapter
Q4B FAQs Frequently Asked Question
Q5A – Q5E Quality of Biotechnological Products
Q5A(R1) Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin
Q5A(R2) EWG Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin
Q5B Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products
Q5C Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products
Q5D Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products
Q5E Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process
Q6A- Q6B Specifications
Q6A Specifications : Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances
Q6B Specifications : Test Procedures and Acceptance Criteria for Biotechnological/Biological Products
Q7 Good Manufacturing Practice
Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Q7 Q&As Questions and Answers: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
Q8 Pharmaceutical Development
Q8(R2) Pharmaceutical Development
Q8/9/10 Q&As (R4) Q8/Q9/Q10 – Implementation
Q9 Quality Risk Management
Q9 Quality Risk Management
Q9(R1) EWG Quality Risk Management
Q8/9/10 Q&As (R4) Q8/Q9/Q10 – Implementation
Q10 Pharmaceutical Quality System
Q10 Pharmaceutical Quality System
Q8/9/10 Q&As (R4) Q8/Q9/Q10 – Implementation
Q11 Development and Manufacture of Drug Substances
Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities)
Q11 Q&As Questions & Answers: Selection and Justification of Starting Materials for the Manufacture of Drug Substances
Q12 Lifecycle Management
Q12 Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management
Q12 IWG Training on Regulatory and Technical Considerations for Pharmaceutical Product Lifecycle Management
Q13 Continuous Manufacturing of Drug Substances and Drug Products
Q13 EWG Continuous Manufacturing of Drug Substances and Drug Products
Q14 Analytical Procedure Development Q2(R2)/Q14 EWG Analytical Procedure Development and Revision of Q2 (R1) Analytical Validation
Reference: ICH.org
References:
1. WHO GMP Guidelines: Guide to Master Formulae, WHO/FWC/IVB/QSS/VQR, 2011
EU and PIC GMP guidelines: EudraLex Volume 4, Chapter 4: Documentation
PIC/S guidelines: Chapter 4: Documentation
Health Canada GMP guidelines: Good manufacturing practices guide for drug products (GUI-0001), Manufacturing control, C.02.011
U.S. FDA: CFR 21, Chapter I, Subchapter F: Biologics, Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals; Subpart F–Production and Process Controls, Sec. 211.100 Written procedures; deviations; and Subpart J–Records and Reports; Sec. 211.186 Master production and control records
U.S. FDA: CFR 21, Chapter I, Subchapter F: Biologics; Subchapter C: Drugs General; Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals; Subpart J– Records and Reports; Sec. 211.188 Batch production and control records.
India: The drugs and cosmetics act, 1940 and The drugs and cosmetics rules, 1945, Schedule M, 12. Documentation and records]
2. PART 314 — APPLICATIONS FOR FDA APPROVAL TO MARKET A NEW DRUG, Subpart A – General Provisions Sec. 314.3 Definitions.
3. 21 CFR PART 210: CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING, PROCESSING, PACKING, OR HOLDING OF DRUGS; GENERAL Sec. 210.1 Status of current good manufacturing practice regulations.
5. Wet Granulation:
End-Point Determination and Scale-Up, By Michael Levin, Ph. D., Metropolitan Computing Corporation East Hanover, New Jersey, USA
6. Saudi Pharmaceutical Journal
Volume 20, Issue 1, January 2012, Pages 9-19, Saudi Pharmaceutical Journal, Review article, Upgrading wet granulation monitoring from hand squeeze test to mixing torque rheometry Author links open overlay panel Walid F. Sakr Mohamed A. Ibrahim Fars K. Alanazi Adel A. Sakr
7. natoli.com
8. pacifictools.in

160+ Pharmaceutical Interview Questions and Answers for Oral Solid Formulations
This page covers most of the interview questions and answers during a technical round in Production Oral Solid Dosage. The interview questions cover questions from basic to advance level of technical aspects. These interview questions and answers will help to crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
The topics covered here are the Granulation process, the Tablet Compression process, the Coating process, and associated topics. In addition, the interview questions and answers cover various equipment used for the manufacturing process of solid oral formulation such as Compression Machine, Coating Machine, Graduation equipment, and supporting accessories.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning, and best of luck with your interview! Happy Learning.
1. What is the granulation end point?
End-point can be defined by the formulator as a target particle size mean or distribution. [Reference: 5]
The agglomerate growth in wet granulation processes depend mainly on rheology of the wet powder mass, as an adequate consistency is necessary for a controllable coalescence and growth of smaller agglomerates into larger agglomerates.[Reference: 6]
2. Method for end point detection for wet granulation process.
i. Banana breaking test or Hand squeeze test
ii. Ampere load method
iii. Measurements of power consumption of the mixer motor
iv. Impeller Torque
v. Binder solution addition and measurement of Loss On Drying (LOD)
vi. Torque value
3. What is the Banana breaking method or Hand squeeze test for determination of granulation end point?
Wear hand gloves, take one handful of wet mass in the palm and press to make a lump. Open the palm and break the lump by pressing the thumb at the center of the lump.
4. What is the principle of metal detectors?
The operation of these metal detectors is based on the principles of electromagnetic induction. Metal detectors contain one or more inductor coils that are used to interact with metallic elements. Metallic contaminant in the product creates a high frequency magnetic field within the detector coil, which in turn activates a reject flap.
5. What is the fail safe mechanism of metal detectors?
When a metal detector gets off because of power failure or inadvertently did not start before the start of the batch, the flap should remain open so that tablets get rejected. It must be challenged at regular intervals (e.g.: start, intermittently and at the end of shift) to make sure they are effective.
6. What are typical standards used for the challenge of metal detectors?
Tablet: Ferrous – 0.3 mm, Non-ferrous – 0.3 mm, Stainless Steel – 0.5 mm, Plain (without metal as a blank)
Capsule: Ferrous – 0.1 mm, Non-ferrous – 0.15 mm, Stainless Steel – 0.2 mm, Plain (without metal as a blank)
7. What is tablet tooling?
Tooling consists of three parts.
i. Upper punch, ii. Lower punch, and iii. Die
8. What is the meaning of one set of tablet tooling?
One set consists of one Upper punch, one Lower punch and a Die.
9. What is a tooling station?
The place where one set accommodates the tablet press.
10. Explain parts of tablet tooling.
Punch terminologies:


Head: Head is the top part of upper and lower punch which contacts the machine’s cams and the pressure rollers apply the pressure to the head to compress the tablets.
Head flat or Dwell Flat: Top flat area of the head. Compression force applied through the upper punch head flat and ejection pressure applied through the head flat of lower punches. Dwell time for compression is determined based on the Head flat hence, it is also called Dwell Flat.
Outside head angle: The area touches the pressure roller during compression operation.
Inside head angle: The part of the punch head helps the upper punch to lift after compression of tablets and helps in pulling down the lower punches after ejection.
Neck: The part between head and barrel. Allow a clear path to the cam.
Barrel: Allows punch to do vertical up and down movement.
Stem: Area from tip end to barrel edge.
Tip: It is the lowest portion of the punch which is responsible for shape, size and profile of the tablet.
Tip face or cup: Outer part of the tip which gives shape to the tablet. The face also will have embossing depending on the need of the tablet requirement.
Tip length: The straight portion of the stem or tip.
Tip straight: The portion of tip towards the face having a higher diameter of the tip.
Tip relief: Tip part other than the tip straight.
Working length: This is the distance from the head flat to bottom of the cup.
Overall length: Length from punch head flat to the tip.
Key or woodruff key: An elevated portion in the middle of the punch barrel. The key with upper punches for shaped tablets to prevent punch rotation when the punch is out of the die.
Types of heads: Domed head and Flat head
Difference between Flat head and Domed head – Domed head punches have smaller flat heads.


The extended head flat offers multiple benefits, including a longer dwell time (the time the head flat spends in contact with the pressure roll) at a given press speed to better compact poorly compressible products. The longer dwell time may even reduce the amount of force required to attain specific tablet hardness.
Whereas, round extended head flats don’t require keyed tooling as oval ones do, and that allows them to be used on multiple makes and models of tablet presses.
Dwell time: It is the time the head flat spends in contact with the pressure roller.
Land: The area between the outer part of the punch cup and the outside edge of the punch tip.
Cup depth: Distance between tip edge to the center point of the cup.
Barrel chamfer: Chamfers at the ends of the punch barrel.
Die terminologies:
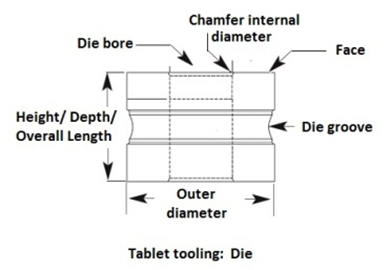
Clearance: pace between die bore diameter and punch tip diameter
Die Bore: It is the cavity of the die where granules are converted into tablets.
Die height or overall length: The height from top to bottom face of the die.
Die chamfer: This is the angle of the die bore at entry point. It guides the upper punch into the die bore.
Die groove and die lock: The radial groove around the die outer diameter requires locking the die into the turret with the help of die lock.
Die taper: Taper is a feature that increases the bore size at the top of the die then nominal bore size. It helps to release air from the die cavity during the compression. The feature helps to reduce tablet defects, such as capping and lamination.
Annealing: This is a heat treatment process to reduce the hardness to make it more workable. The processing done fragile punch tips to decrease the hardness of the punch cups which helps in reducing punch tip fracturing.
11. Describe tablet tooling standards?
There are two types of standards for tablet tooling:
i. US specification provided by Tableting Specification Manual (TSM) (This is the only tooling specifications are the only published standards for the tablet compression industry). The standard is established by the American Pharmacists Association (APhA),
ii. European standard known as the EU, or “Euronorm” standard.
EU, or Euronorm standard tool configurations are not published or governed by an organization or association. The EU standard is the most common tooling configuration used outside the U.S. [Reference: 7]
12. Differences between TSM and EU Tooling Configurations
| Tableting Specification Manual (TSM) Tooling Configurations | EU Tooling Configurations |
| Angled top profile | Domed head profile |
| Inside head angle for “B” punches is 37° | Inside head angle for “B” punches is 30° |
| Overall head thickness is greater in both “B” and “D” | Overall head thickness is lesser in both “B” and “D” |
| Overall punch length of the TSM tool is 0.010 inches shorter than the EU | Overall punch length of the EU tool is 0.010 inches longer than the TSM |
13. What is the type of tablet tooling for a compression machine with die and punch dimension?
There are the following six types of tablet tooling and its dimensions.
Tooling specification | Nominal punch barrel diameter (mm) | Nominal punch die diameter (mm) | Maximum tablet diameter for round tablet (mm) | Maximum tablet diameter for shaped tablet (mm) |
| TSM or EU B | 19 | 30.16 | 16 | 16 |
| TSM or EU D | 25.35 | 38.1 | 25 | 25 |
| TSM or EU BB | 19 | 24 | 13 | 14 |
| TSM or EU DB | 25.35 | 30.16 | 19 | 19 |
| TSM or EU BBS | 19 | 21 | 12 | 13 |
| TSM or EU A | 12.7 | 17 | 8 | 8 |
14. What is the choice for material of construction (MoC) of Die and Punch?
i. OHNS (T) Oil Hardened Non shrinking steel (Tungsten) – AISI 01 (American Iron and Steel Institute)
ii. HCHC – High carbon High chromium steel – AISI D3
15. What is the choice for material of construction (MoC) of Turret?
Three piece turret having the center die table of SS 316 and the Upper and lower piece of Spheroidal Graphite Iron (SG Iron) with ELNP (ELECTROLESS NICKEL PLATING) Coating.
16. What is the chemical composition of OHNS steel?
| Element | Content (%) |
| C | 0.85-1.00 |
| Si | 0.15-0.35 |
| Mn | 1.00-1.20 |
| P | 0.03 Max |
| S | 0.03 Max |
| Cr | 0.50-0.70 |
| W | 0.50-0.70 |
17. How much is the lifespan of the die and punches?
The life span of punches and dies is purely depending on materials of construction (MoC) as well as usage and handling.
OHNS Punches and HCHC Dies: 4 million tablets
HCHC Punches and HCHC Dies: 8 Million tablets Complete hard chrome plating punches and
HCHC dies: 10 Million tablets
18. How to maintain Punches and Dies?
The maintenance of compression tooling becomes easy by following these mentioned steps:
Step 1: Cleaning during every product change over or on a periodic basis. They can be wiped with IPA and dried using a lint free cloth.
Step 2: Periodic evaluation to increase their shelf life. Industry practice is to
Step 3: The regular visual inspection of the tooling should be done after each cleaning to check for physical damages to head, tip, die, and embossing, debossing using Magnifying glass.
Step 4: Periodic inspection of the punch set should be done using measuring tools. Frequency for die and punch inspection should be such that first inspection after procurement should be done after 3 million tablet compression using each tool set and subsequently after each million.
Step 5: Compression punch set should be lubricated to decrease friction and enhance the operational activity of the tablet compression machine. A non-toxic, FDA approved oils and greases should be used for preservation and lubrication purposes. Storage of tooling should be done in the safe and moisture-free place.
19. What should be available in a punch set/ tablet tooling inspection kit?
| 1 | Dial gauge compactor stand with a least count of 0.001 inch |
| 2 | Micrometer Range from 0.25 mm with least count : 0.01 mm |
| 3 | Punch holding bushes for B type punch and D type of punch |
| 4 | Punch height gauge |
| 5 | Die Outer diameter block for all types of die i.e. D, B, BB, DB etc. |
| 6 | Magnetic V – block |
| 7 | GO – NO GO gauges |
| 8 | Magnifying Glass |
| 9 | Die Pocket Cleaner |
20. Explain about the punch set/ tablet tooling inspection program?
i. Height uniformity of the punches
ii. Punch body to punch tip concentricity
iii. Die bore Go / No go status
iv. Other dimensions as per the drawing
v. Punch tip to die before clearance
vi. Hollow penetration of the punches
vii. Die outdoor diameter consistency
viii. Die height regularity
ix. Die hole GO and NO GO examination
x. Die internal diameter to outer diameter concentricity
21. Issue that may arise after prolonged use for punch set/ tablet tooling.
i. Tablet weight difference
ii. Tablet hardness difference
iii. Powder leakage from lower punch or collar formation
iv. Twisting of small size punch tips
22. Commonly used material of construction in the pharmaceutical industry for equipment manufacturing?
i. Commonly used materials of construction in the pharmaceutical industry for product contact parts are Stainless steel 316, 316 L, Toughened glass, Silicon, Food grade Teflon etc.
ii. Commonly used materials of construction in the pharmaceutical industry for non-product contact parts are Stainless steel 304, 304 L, Teflon, Anodized or powder coated aluminum, brass etc.
23. Explain characteristics of different grades of stainless steel material.
Following are the different types of types of Stainless and similar material with its characteristics.
| Stainless and similar material | Application for the different types of environment |
| Hastelloy C276 | Excellent corrosion resistance in a wide range of severe environments |
| 316, 316L | Chemical corrosion |
| 304, Custom 450 | Medium Corrosion |
| 430, 431, Custom 455 | Industry polluted air |
| 405, 410, 420, 440 | Clean air |
24. Explain most commonly used stainless steel for pharmaceutical industry and its attributes.
Following most commonly used stainless steel for the pharmaceutical industry and its attributes.
| Commonly known names | UNS | EN | Attributes |
| 304 | S30400 | 1.4301 | Good at general corrosion resistance to acidic as well as caustic material |
| 304L | S30403 | 1.4307 | Good at general corrosion resistance to acidic as well as caustic material with low carbon and improved performance |
| 316 | S31600 | 1.4401/ 1.4436 | Improved corrosion resistance to most acidic as well as caustic material with high temperature or chloride present |
| 316L | S31603 | 1.4404/ 1.4432 | Improved corrosion resistance to most acidic as well as caustic material with high temperature or chloride present with low carbon and improved performance |
25. How to distinguish SS 202, SS 304 and SS 316 grade stainless steel?
Using molybdenum detection electrolyte kit. To know how to perform the test, Click Here.
26. What is the composition of SS 304, SS 304L, SS 316, and SS 316L?
Following are the composition of SS 304, SS 304L, SS 316, and SS 316L

27. What is the meaning of Suffix L in SS 316L and SS 304L?
Suffix L for SS 316 and SS 304 represents low carbon content. Standard grade consists of ≤ 0.08 % carbon and low carbon grade consists of ≤ 0.03 % carbon content.
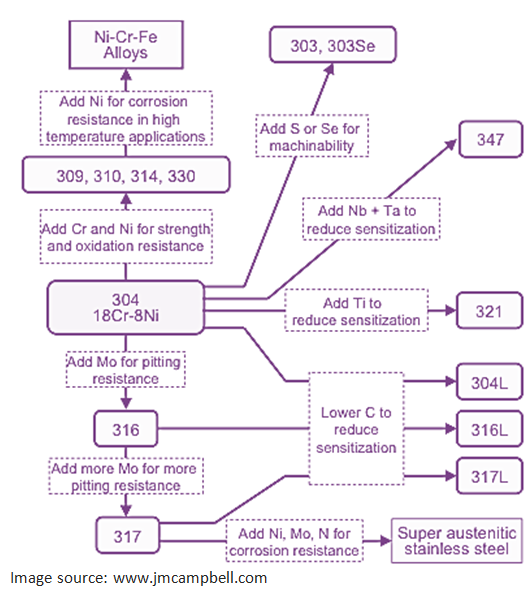
28. What are the different types of stainless steel in terms of material composition?
29. What is Hastelloy?
Hastelloy is a nickel-molybdenum-chromium superalloy with an addition of tungsten designed to have excellent corrosion resistance in a wide range of severe environments. The high nickel and molybdenum contents make the nickel steel alloy especially resistant to pitting and crevice corrosion in reducing environments while chromium conveys resistance to oxidizing media. The low carbon content minimizes carbide precipitation during welding to maintain corrosion resistance in as-welded structures. This nickel alloy is resistant to the formation of grain boundary precipitates in the weld heat-affected zone, thus making it suitable for most chemical process application in an as welded condition.
Source: megamex.com
30. In what applications is Hastelloy used?
i. Pharmaceutical and food processing equipment
ii. Chemical processing components like heat exchangers, reaction vessels, evaporators, and transfer piping
iii. Sour gas wells
iv. Waste treatment
31. Which are typical in-process control tests for tablets?
Dimensions
Hardness
Friability
Thickness
Disintegration
Weight variation
Content uniformity
Dissolution
Leakage testing for strip and blister packaging
Checking printing matter during packaging
Physical appearance of packs
32. Explain Disintegration time (DT) of different types of tablets?
Disintegration time (DT) of different types of tablets is as follows.
| Types of tablets | Disintegration time (DT) |
| Uncoated | 15 minutes |
| Plain coated tablet | 60 minutes |
| Enteric coated tablet | 3 hours |
| Dispersible tablet | 3 minutes |
| Effervescent tablet | ˂ 3 minutes |
| Sublingual tablet | 4 hours |
| Buccal tablet | 4 hours |
| Vaginal tablet | 60 minutes |
| Chewable tablet | not required |
33. What are the types of hardness testers?
Manual hardness tester
Monsanto tester

Pfizer tester

Strong-cobb

Automatic hardness testers
Erweka tester

Dr. Schleuniger tester

34. Which instruments are used for testing tablet dimension?
A. Vernier caliper

1. Outside jaws: used to measure external length
2. Inside jaws: used to measure internal length
3. Depth probe: used to measure depth
4. Main scale (cm)
5. Main scale (inch)
6. Vernier (cm)
7. Vernier (inch)
8. Retainer: used to block/release movable part
B. Micrometer screw gauge

35. Which instruments are used for testing of tablet friability?
Roche friabilator is used for testing of tablet friability.
36. Explain the construction of tablet friability tester, testing process and limit.
i. Plastic chamber that revolves at 25 rpm
ii. For testing it operates for 100 revolutions
iii. Dropping the tablets from a Distance of 6 inches in the tester
iv. The tablets are weighed before and after rotation. De-dust tablets after rotation. Perform calculation in percentage weight loss.
v. Limit of friability testing is NMT 1.0 %
37. What is Disintegration Time (DT) Test?
Disintegration time is time taken to break down the tablets into granules or primary powder particles.
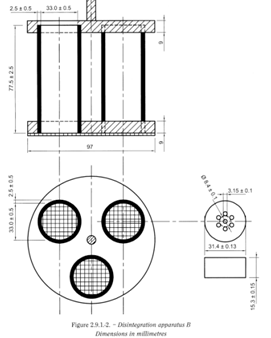
38. Explain the tablet disintegration tester parts.
i. Basket rack assembly
ii. A suitable vessel for the immersion liquid
iii. Heater for fluid heating between 35°C and 39°C
iv. Basket in the immersion in fluid at frequency rate between 28 and 32 cpm (Cycle per minute)
v. Distance of up and down not less than 5 cm and not more than 6 cm


39. What are the common tablet defects at compression stage?
Weight variation
High friability
High or low hardness (resistance to crushing)
Sticking
Picking
Capping
Laminating
Chipping
Mottling
Double press or impression
Cracking
Binding
Edging or Flashing of tablet
Disintegration time abnormality
Black Spot/ particles/ fiber
Improper embossing or debossing
Layer separation
Improper Layers
40. What are the different types of tablet coating defects?
Blistering
Chipping
Cratering
Sticking or Picking
Pitting
Blooming
Blushing
Color Variation
Infilling
Orange Peel or Roughness
Cracking or Splitting
Twinning Erosion
Bridging
41. Definitions of tablet defects at compression stage.
Weight variation: Individual weight of tablet outside of the accepted criteria.
High friability: The condition of tablets being friable. The tendency of tablets to break into smaller pieces or weight loss of powder from the surface of the tablets due to mechanical action such as transportation conditions. When percentage loss is more than 1%, it is considered as high friability.
To view images of tablet defects and to download high resolution poster, CLICK HERE
High or low hardness (resistance to crushing): Individual hardness of tablets is outside the accepted limit (upper limit or lower limit). Hardness limits are determined during the development phase of the product.
Sticking: Sticking is a defect of the tablet wherein the portion tablet surface sticks to the face of the punch or to the die wall during tablet compression activity.
Picking: Picking is a counter part of the sticking. When part of the tablet surface sticks to the punch or to the die wall, the produced tablets are with a pitted surface instead of a smooth surface.
Capping: Capping (or splitting) is a term used to describe the partial or complete separation of the upper or lower segment of the tablet horizontally from the main body of a tablet during ejection from the tablet press.
Laminating: Laminating of tablets means separation of a tablet into two or more different parallel layers.
Chipping: Chipping of tablets is the breaking of tablet edges during manufacturing or handling.
Mottling: Mottling of tablet refers to unequal or nonuniform distribution of color on the tablet surface.
Double press or impression: Double impression is a tablet defect where an embossing or break or score line appears two times on the tablet surface.
Cracking: Cracking of tablets means small fine cracks on the upper and lower central surface of the tablets and sometimes seen on the side wall.
Binding: The term binding is used when tablets stick to the die and do not eject properly out of the die.
Edging or Flashing of tablet: Edging or Flashing means observation of burrs or sharp edges on the edges of tablet.
Disintegration time abnormality – Too fast or too slow disintegration of tablets.
Black Spot/ particles/ fiber – Observation of black spot or particles or fiber which could be indigenous or foreign matter or contaminant.
Improper embossing or debossing: The defect because of improper embossing or debossing of letter, logo or monogram.
Layer separation: Layer separation is the defect of a tablet where layers of tablet get separated. This is usually seen in tablets such as bilayer tablets.
Improper Layers: Improper Layers mainly appear in layered tablets such as bilayer tablets. In this type of defect, the layer does not distinguish sharply.
42. What are the different types of tablet coating defects?
Blistering: Blistering means blistering of a surface film appears when film becomes detached from the tablet’s substrate.
Chipping: Chipping means dented and chipped film mostly on the edges of the tablet.
Cratering: Cratering means volcanic-like cratering happens exposing the tablet surface.
Sticking or Picking: Sticking or picking means sticking of film with each other or with the coating pan resulting in some of the tablet pieces being detached from the core.
Pitting: Pitting is the deformation of the core of the tablet without any visible signs of disruption of the film coating.
Blooming: Blooming is the fading or dulling of a tablet color after it is stored for a prolonged period at a high temperature.
Blushing: Blushing is described as the appearance of white specks or a haziness in the film.
Color Variation: Color variation of tablet film.
Infilling: This refers to the filling of intagliations – i.e., the distinctive words or symbols formed on the tablet.
Orange Peel or Roughness: When film having a rough surface or having a matt surface rather than glossy texture. It appears the same as an orange.
Cracking or Splitting: The cracking defect is observed when the film coating of the tablet cracks in the crown area or splits around the edges.
Twinning: Twinning is when two tablets stick together.
Erosion: Coating gets removed from the surface due to friction between tablets.
Bridging: When coating material fills in the logo or letter, bridging occurs.
43. Explain the remediation for various types of tablet defects.

44. What are the different sizes of hard gelatin capsules?
The capsule sizes are 000, 00, 0, 1, 2, 3, 4, and 5 from the largest to the smallest. There are few special sizes of capsules available such as 2el, 1el, 0el, 0el+, 0xel, 00el etc. Details are tabulated below.


45. What are the US Mesh size and corresponding micron, mm and inch for commonly used sieves, provide a few examples?
| U.S. Mesh | Microns | Inches | Millimeters |
| 3 | 6730 | 0.2650 | 6.730 |
| 4 | 4760 | 0.1879 | 4.760 |
| 5 | 4000 | 0.1570 | 4.000 |
| 6 | 3360 | 0.1320 | 3.360 |
| 7 | 2830 | 0.1110 | 2.830 |
| 8 | 2380 | 0.0937 | 2.380 |
| 10 | 2000 | 0.0787 | 2.000 |
| 12 | 1680 | 0.0661 | 1.680 |
| 14 | 1410 | 0.0555 | 1.410 |
| 16 | 1190 | 0.0469 | 1.190 |
| 18 | 1000 | 0.0394 | 1.000 |
| 20 | 840 | 0.0311 | 0.841 |
| 25 | 707 | 0.0280 | 0.707 |
| 30 | 595 | 0.0232 | 0.595 |
| 35 | 500 | 0.0197 | 0.500 |
| 40 | 400 | 0.0165 | 0.400 |
| 45 | 354 | 0.0138 | 0.354 |
| 50 | 297 | 0.0117 | 0.297 |
| 60 | 250 | 0.0098 | 0.250 |
| 70 | 210 | 0.0083 | 0.210 |
| 80 | 177 | 0.007 | 0.177 |
| 100 | 149 | 0.0059 | 0.149 |
| 120 | 125 | 0.0049 | 0.125 |
| 140 | 105 | 0.0041 | 0.105 |
| 170 | 88 | 0.0035 | 0.088 |
| 200 | 74 | 0.0029 | 0.074 |
| 230 | 63 | 0.0024 | 0.063 |
| 270 | 53 | 0.0021 | 0.053 |
| 325 | 44 | 0.0017 | 0.044 |
| 400 | 37 | 0.0015 | 0.037 |
| 450 | 32 | 0.0013 | 0.032 |
| 500 | 25 | 0.0010 | 0.025 |
| 550 | 25 | 0.0009 | 0.023 |
| 635 | 20 | 0.0008 | 0.020 |
| 800 | 15 | 0.0006 | 0.015 |
| 1250 | 10 | 0.0004 | 0.010 |
Reference: ASTM E11
46. Give examples of sieve size and corresponding number of Apertures per Linear inch.
| Sr. No. | Sieve Size | Number of Apertures per Linear Inch |
| 1 | 10 # | 9-11 |
| 2 | 12# | 11-13 |
| 3 | 14# | 13-15 |
| 4 | 16# | 15-17 |
| 5 | 20# | 19-21 |
| 6 | 24# | 23-25 |
| 7 | 30# | 28-32 |
| 8 | 40# | 38-42 |
| 9 | 60# | 57-63 |
| 10 | 80# | 77-83 |
| 11 | 100# | 97-103 |
47. What is the aperture size of the screen?
The size of a square opening (length of clear space between individual wires) is called the aperture size of the screen.
48. What is the Mesh number of screens?
Mesh number of screens is defined as the number of aperture or opening per linear inch of the screen. E.g. A screen having 10 square openings per inch is said to have Mesh no. 10. Higher the mesh number, smaller the aperture size of the screen.
49. What is the broad classification of Granulation techniques?
Granulation technique is broadly classified into two types as follows:
(i) Dry granulation
a. Slugging technique
b. Roller compaction
c. Pneumatic dry granulation
(ii) Wet granulation
a. Steam granulation
b. Reverse wet granulation
c. Moisture-Activated Dry Granulation
d. Thermal adhesion granulation
e. Melt granulation
f. Freeze granulation
g. Foam granulation
50. What are the equipment used during the Wet Granulation Process?
Depending on process, batch size and type of process steps, following equipment are used during the Wet Granulation Process.
Low Shear mixer/ granulator
High Shear mixer/ granulator OR Rapid Mixture granulator
Fluid-Bed granulator/ dryer,
Spray Dryer,
Extruders and Spheronizer
Vibratory sifter
Cutting mills such as Multimill, Co-mill, Pin Mill etc.
Hammer mill
51. What are the equipment used during the Dry Granulation Process?
Depending on process, batch size and type of process steps, following equipment are used during the Dry Granulation Process.
Role com
Low Shear mixer/ granulator
High Shear mixer/ granulator OR Rapid Mixture granulator
Vibratory sifter
Cutting mills such as Multimill, Co-mill, Pin Mill etc.
Roller Compactor Granulator
Compression machine to generate slugs
52. What is the most commonly used testing method to determine powder flow?
There are four commonly used methods for testing powder flow:
(1) Angle of repose
(2) Compressibility index or Hausner ratio
(3) Flow rate through an orifice
(4) Shear cell
Reference: General Chapters: <1174> POWDER FLOW
53. What is Angle of Repose?
Definition of Angle of Repose: The angle of repose is a relatively simple technique for estimating the flow properties of a powder. It is determined by allowing a powder to flow through a funnel and fall freely onto a fixed diameter base. The height and diameter of the resulting cone are measured. For further reading on Angle of Repose and download free excel, click on this link.
54. What is the Compressibility index and what is Hausner Ratio?
Definition of Compressibility index: The Compressibility Index (Carr Compressibility Index) is a measure of the tendency of a powder to be compressed. It is a measure of the powder’s ability to settle, and it permits an assessment of the relative importance of interparticulate interactions.
Definition of Hausner Ratio: The Hausner Ratio is measures that can be used to predict the tendency of a given powder sample to be compressed. Hausner Ratio reflects the importance of interparticulate interactions.
55. What is the disintegration time of Chewable Tablets?
Disintegration time test is not applicable for chewable tablets.
56. What is the standard number of rotations used for friability test?
100 Revolutions / 4 Minutes, 25 rotations per minute
57. What is the fall height of the tablets in the friabilator during friability testing?
Fall height of the tablets is in the friabilator is 154.0 – 158.0 mm (156.0 ± 2.0 mm)
58. Which capsule is bigger in size – size ‘0’ or size ‘1’?
‘0’ size
59. How many tablets shall be taken for checking friability?
6.5 g for the tablets with unit mass equal or less than 650 mg. For tablets with unit mass more than 650 mg, it requires 10 tablets.
60. What is the mesh aperture of DT apparatus?
Mesh aperture of DT apparatus is #10. That is 1.8 -2.2 mm.
61. What is the standard frequency of upward and downward movement of a basket-rack assembly in a DT apparatus?
28 – 32 cycles/minute
62. What are the parameters tested during the calibrating DT apparatus?
Parameters covered during the DT apparatus calibration are as follows
(1) Number of strokes per minute (Limit: 29-32 cycles/min)
(2) Temperature by probe and standard thermometer (Limit: 37 ± 1 °C). (3) Distance travelled by basket (Limit: 53 -57 mm)
63. What is the Disintegration time for different types of tablets?
| Type of Tablets | Disintegration time |
| Uncoated Tablets | 15 minutes |
| Film-coated | 30 minutes |
| Other coated tablets | 60 minutes |
| Enteric-coated Tablets | |
| 0.1M hydrochloric acid | Should not disintegrate in 120 minutes |
| Phosphate buffer pH 6.8, | 60 minutes. |
| Dispersible and Soluble Tablets | within 3 minutes |
| Effervescent Tablets | 5 minutes |
| Chewable tablets | Not applicable |
64. What are the different clean room conditions? Explain it.
There are three conditions of clean rooms – as built, at rest and in-operation.
As built:

Condition where the installation is complete with all services connected and functioning but with no production equipment, materials, or personnel present.
At rest:
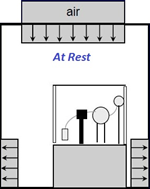
Condition where the installation is complete with equipment installed and operating in a manner agreed upon by the customer and supplier, but with no personnel present.
In operational:

This condition relates to carrying out room classification tests with the normal production process with equipment in operation, and the normal staff present in the room.
Reference: WHO Working document QAS/02.048/Rev.2
65. What are the different types of air locks? Explain it.
There are three types of air locks, Cascade airlock, Sink airlock and Bubble airlock.
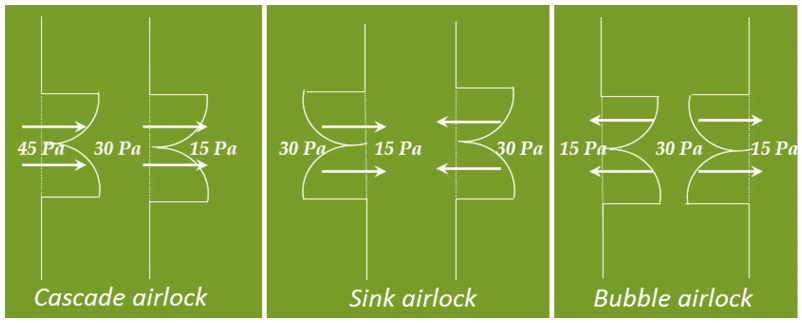
Cascade airlock: high pressure on one side of the airlock and low pressure on the other side.
Bubble Airlock: high pressure inside the airlock and low pressure on both outer sides.
Sink Airlock: low pressure inside the airlock and high pressure on both outer sides.
Reference: WHO Working document QAS/02.048/Rev.2
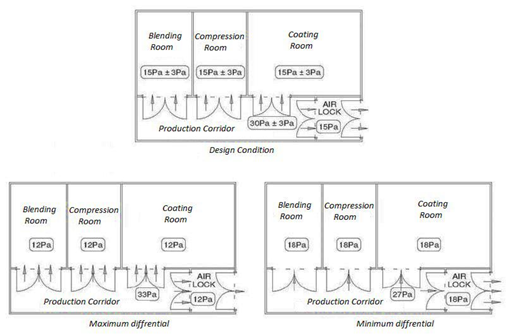
66. What should be the differential pressure between two adjacent clean room zones?
The most widely accepted pressure differential to achieve containment between the two adjacent zones is 15 Pa, but pressure differentials of between 5 Pa and 20 Pa could be acceptable.
Where the design pressure differential is too low and tolerances are at opposite extremities, a flow reversal can take place. E.g. where a control tolerance of ±3 Pa is specified, the implications of the upper and lower tolerances on containment should be evaluated.
67. Comparison of pascals to mm of water?
Pascals to Millimeters of water formula
mm H2O =Pa * 0.10197
Pascals (Pa)
Millimeters of water (mm H2O)
| Pascals | Millimeters of water | Pascals | Millimeters of water | Pascals | Millimeters of water |
| 1Pa | 0.10mm H2O | 16Pa | 1.63mm H2O | 31Pa | 3.16mm H2O |
| 2Pa | 0.20mm H2O | 17Pa | 1.73mm H2O | 32Pa | 3.26mm H2O |
| 3Pa | 0.31mm H2O | 18Pa | 1.84mm H2O | 33Pa | 3.37mm H2O |
| 4Pa | 0.41mm H2O | 19Pa | 1.94mm H2O | 34Pa | 3.47mm H2O |
| 5Pa | 0.51mm H2O | 20Pa | 2.04mm H2O | 35Pa | 3.57mm H2O |
| 6Pa | 0.61mm H2O | 21Pa | 2.14mm H2O | 36Pa | 3.67mm H2O |
| 7Pa | 0.71mm H2O | 22Pa | 2.24mm H2O | 37Pa | 3.77mm H2O |
| 8Pa | 0.82mm H2O | 23Pa | 2.35mm H2O | 38Pa | 3.87mm H2O |
| 9Pa | 0.92mm H2O | 24Pa | 2.45mm H2O | 39Pa | 3.98mm H2O |
| 10Pa | 1.02mm H2O | 25Pa | 2.55mm H2O | 40Pa | 4.08mm H2O |
| 11Pa | 1.12mm H2O | 26Pa | 2.65mm H2O | 41Pa | 4.18mm H2O |
| 12Pa | 1.22mm H2O | 27Pa | 2.75mm H2O | 42Pa | 4.28mm H2O |
| 13Pa | 1.33mm H2O | 28Pa | 2.86mm H2O | 43Pa | 4.38mm H2O |
| 14Pa | 1.43mm H2O | 29Pa | 2.96mm H2O | 44Pa | 4.49mm H2O |
| 15Pa | 1.53mm H2O | 30Pa | 3.06mm H2O | 45Pa | 4.59mm H2O |
68. What is the difference between disintegration and dissolution?
Disintegration is a process of breaking down of granules into small fragments.
Dissolution is the process in which a substance goes into a solution. It is a measure of bioavailability and therapeutic effectiveness. Dissolution is also called drug release.
69. What is the purpose of maintaining pressure gradients between processing areas and service corridors?
Pressure gradients are maintained to avoid cross contamination of products through air.
70. In oral solid manufacturing facility ‘positive’ pressure is maintained in processing area or in service corridors?
Positive pressure is maintained in service corridors with respect to processing rooms.
71. What should be the characteristic of Finger Bags used for Fluid Bed Dryer (FBD) or Fluid Bed Processor (FBP)?
Antistatic
Waterproof cloth
Able to handle granulation of highly viscous materials, such as extract powder
Non-stick cloth bag
Good permeability
Good boiling effect
Cost-efficient and durable
72. What is generally used material of construction of Finger Bags used for Fluid Bed Dryer (FBD) or Fluid Bed Processor (FBP)
Finger bags are constructed by a combination of Antistatic polymer, Nylon fabric and Cotton fabric. Following materials are also used for Finger bags’ construction.
Antistatic
Epitropic
Polypropylene
Polyester
Stain
73. What are the typically seen micron ratings of Finger Bags used for Fluid Bed Dryer (FBD) or Fluid Bed Processor (FBP)?
Micron Rating: 1, 5, 10, 15, 25, 50, 100
74. What is the formula for calculating friability test results?
Friability (%) = Weight 1 – Weight 2 / Weight 1 X 100
Where,
Weight 1 = Weight of Tablets (Initial / Before Tumbling) & Weight 2 = Weight of Tablets (After Tumbling or friability)
75. What is the limit of friability test results?
Limit: Friability (%) = Not More Than 1.0 %
76. What is Rheology?
Rheology (Greek words rheos meaning flow and logos meaning science) is the study of the flow under the influence of stress. The principle can be applied to solids, liquids, and gases.
77. Explain the types of excipients.
Organoleptic
o Color
o Flavor
o Sweetener
Stabilizers
o Preservative
o Antioxidant
o Emulsifier
o Suspending Agent
Dose Accuracy
o Diluent
o Bulking agent
o Filler
Process Aids
o Binder
o Lubricant
o Glidant
o Anti-adherent
Drug release
o Disintigrant
o Penetration enhancer
o Coating agent
78 What are the types of tablets? Or Classification of Tablets.
Classification of tablets by route of administration
Oral tablets for ingestion
Implantation tablets
Chewable tablets
Tablets used in the oral cavity
Buccal and sublingual tablets
Dental cones
Vaginal tablets
Tablets used to prepare solutions
Effervescent tablets
Dispensing tablets (DT)
Hypodermic tablets (HT)
Tablet triturates (TT)
Troches and lozenges
Classification of tablets by manufacturing process
Compressed tablets
Layered tablets
Sugar coated tablets
Film-coated tablets
Classification of tablets by onset of action
Immediate release tablets
Repeat-action tablets
Delayed-action and enteric coated tablets Controlled release tablets
79. What are the various types of tablets?
Following are various types of tablets.
- Compressed Tablet (CT)
- Sugar-Coated Tablets (SCT)
- Film-Coated Tablets (FCT)
- Enteric-Coated Tablets (ECT)
- Multiple Compressed Tablets (MCT)
- Layered Tablets
- Press-Coated Tablets
- Controlled-Release Tablets (CRT)
- Tablets for Solution (CTS)
- Effervescent Tablets
- Compressed Suppositories or Inserts
- Buccal and Sublingual Tablets
- Molded Tablets or Tablet Triturates (TT)
- Dispensing Tablets (DT)
- Hypodermic Tablets (BT)
- Compressed Tablets (CT)
80. What are the ingredients used to formulate or manufacture the various types of tablets?
Following are ingredients used to formulate or manufacture the various types of tablets.
- Active pharmaceutical ingredient
- Diluents
- Binders
- Lubricants
- Glidants
- Disintegrants
- Coloring Agents
- Flavoring Agents
81. Explain parts of the compression machine.
- Machine Hopper
- Feeder System (force feeder/ gravity feeder)
- Feeder housing
- Feed pedals
- Punches System
- Upper punch system
- Lower punch system
- Die System
- Turret
- Machine Cam Tracks
- Powder Filling Station and Weight Control
- Compression Rollers (Pre-compression, main-compression)
- Tablet Press Ejection Cam
- Take –off blade
- Discharge Chute
- Touch Screen Control Panel (HMI/ MMI)
- Electric Motors, Gears and Belts
- Lubrication Systems
82. What are the types of tablet press or compression machines?
Broadly, we there are two main types of tableting machines:
- Single-station tablet press or compression
- Multi-station tablet press or compression
- Single rotary machine
- Double rotary machine
83. What are the types of tablet Coating Machines available in the market?
• Standard Coating Pan
• Perforated Coating Pan
• Fluidized Bed Coater
84. What are the advantages of the tablet coating process?
Following are the advantages of tablet coating:
• It enhances its appearance
• Easy to consume
• Minimizes the unpleasant colour, odour or taste of drug
• Identification
• Helps to reduces drug degradation by protecting it from environmental factors
• Facilitates the packing process
• Functional coating gives functional advantages like extended release, enteric release, prolonged release etc.
85. What are the disadvantages of the tablet coating process?
Following are the disadvantages of tablet coating:
• Cost of operation, resources and material
• Equipment operation and maintenance cost
• May change tablet properties
• Residual solvent toxicity
• Waiting time between processes
• Increase process lead time
• Increase analysis load
86. Explain parts of tablet coating equipment or machine.
Following are the parts of tablet coating equipment or machine
• Perforated coating pan
• Baffles in coating pan
• Spray gun
• Spray pump e.g. Peristalsis pump, Diaphragm pump
• Electric Motors
• Air Handling unit for inlet air
o Inlet air blower
o Air Handling unit housing
o Primary filter (Washable)
o High temperature HEPA filter
o Inlet air temperature sensor
o Differential air pressure sensors across the above filters
• Ducting to supply air and exhaust air
• Control panel with PLC Control and HMI
• Exhaust air blower
• Dust collector or air Scrubber (Wet or Dry)
• Solution preparation tank
87. Explain parts of Fluid bed dryer and Fluid bed processor?
Following are the parts of Fluid bed dryer and Fluid bed equipment
• Plenum
• Product Container with PU wheel trolley
• Inflatable gasket for connection between plenum and Product Container, and Product Container and expansion chamber
• Product temperature sensor
• Inlet air temperature and RH sensor
• Damper for inlet air
• Sample collection port in Product Container
• Glass view in Product Container
• Silicon molded Dutch Woven sieves
• Conidur mesh for mechanism of vortex formation
• Expansion chamber
• Glass view in Expansion chamber
• Finger bag
• Finger bag mounting assembly
• Finger bag shaking assembly
• Inflatable gasket to seal finger bag assembly
• Broken Bag Detector (BBD) sensor OR Solid Flow Monitor (SFM) sensor
• Exhaust blower
• Exhaust filters (10 and 3 µm porosity)
• Explosion flap and Explosion port
• Inlet Air Handling Unit
o Air Handling unit housing
o Primary filter (Washable)
o High temperature HEPA filter
o Dew point sensor
o Differential air pressure sensors across the above filters
o Damper
• CFM or Air flow sensor
• Inlet air duct
• Clean in place system
• Differential pressure sensors, across filters, across finger bag, across product container
• Exhaust air temperature
• Spraying system (for top spray granulation)
88. What is a Tablet Deduster?
Deduster is an equipment used in the pharmaceutical and nutraceutical industry to remove surface dust from the tablets.
89. What are different types of tablet Deduster?
- Rotary Vibrating Deduster
- Uphill Deduster
- Brush Type Deduster
- Horizontal Deduster
- Vertical Deduster
i. Vertical Downward Conveying Deduster
ii. Vertical Upward Conveying Deduster
90. Explain in detail about different types of Tablet Deduster.
There are different types of tablet dedusters. Each has its advantages and disadvantages.
a. Rotary Vibrating Tablet Deduster
Rotary Vibrating Tablet Deduster depends on rotation and vibration principles.
It has a vibrated helical path with a perforated sieve. As the tablets vibrate and spin along the helical path with a perforated plate, burrs and dust are wiped from their surface. A dust extraction mechanism extracts the dust and the tablets drop into the collection container.
b. Uphill Tablet Deduster
Uphill tablet Dedusting machine depends on the vibration that removes burr and dust as it elevates the tablets. The deduster elevates and dedusts concurrently.
c. Brush Type Tablet Deduster
It consists of a helically wound brush installed within a stainless steel tube (same as screw conveyor) steered by an adjustable speed motor.
The brush pushes the tablets across the tube till they get to the discharge of the deduster at the top or end.
d. Horizontal Tablet Deduster
These types of tablet dedusters use perforated tubes or perforated stainless steel plates that vibrate from one side to another. It is horizontal but slants downwards that the tablets fall as the vibration conveys them over a distance of around 1 meter.
e. Vertical Tablet Deduster
Vertical tablet dedusters are of two types, Downward Conveying Tablet Dedusters and Upward Conveying Tablet Dedusters. The working principle of both types of dedusters are similar.
The dedusters rely on spiral punctured trays on an anchored central column.
91. Which Material is used to make Tablet Dedusters?
Stainless Steel: Stainless steel forms the largest portion of tablet dedusters.
Acrylic: To make the windows.
92. Which Quality Standards should a Tablet Deduster Comply with?
a. Current good manufacturing practices (cGMP) quality standards
b. ISO certification quality standards
c. CCC compliant
d. CE certification
93. How do you select the tablet deduster?
a. Height of compression machine and containers
b. Tablet Size
c. Output
d. Tablet Hardness and Features of Dust
e. Inclusion of Extra Components such as metal detector
94. Explain parts of multimill?
Material Charging Hopper: To load the material to be milled
Milling chamber: Milling of material happens within the chamber with the help of Cutting blades and desired size of particles are passed through specific size screen
Discharge port: Milled material is discharged from this part
Castor wheels: To move the mill from one place to another place
Operating Panel: To operate the equipment
Screen: To produce desired particle size
Cutting blades: To mill the material
95. What is the Material Of Construction (MOC) of multimill parts?
Product contact parts are made up of SS 316 or SS316L
Non-product contact parts are made up of SS 304 Castor wheels are made up of Polyurethane
96. What is the mechanism of size reduction when using multimill?
a. Impact Milling: Particles are reduced in size by high-speed mechanical impact or impact with other particles; also known as milling, pulverizing, or comminuting.
b. Cutting: Particles are reduced in size by mechanical shearing.
c. Screening: Particles are reduced in size by mechanically induced attrition through a screen. This process commonly is referred to as milling or deagglomeration.
97. What are the typical speeds of multimill?
Fast speed: 2880
Medium fast speed: 2160
Medium speed: 1440
Low speed: 720
Note: Nowadays multimill are available with variable frequency drive where speed can be set as per the requirement (Custom speed).
98. What are the typical settings for milling operation using multimill?
Blade direction: Impact forward or Knife forward
Machine speed: Using V belt adjustment or using VFD as per machine design
99. What is Vibro sifter?
Vibro sifter is equipment used for separating particles based upon particle size alone and without any significant particle size reduction. This process commonly is referred to as screening or bolting. Vibration and Gyratory motion plays an important role to facilitate the screening process.
100. What is the principle of Vibro sifter?
Vibro sifters work on the principle of separation. Particles are segregated based upon particle size alone and without any significant particle size reduction. This process commonly is referred to as screening or bolting. Vibration and Gyratory motion plays an important role to facilitate the screening process.
The mechanism on which Vibro Sifter works is the principle of gyratory vibrations. The material is separated based on its particle size. Once the motor gets energized, vibration is caused in the screen/. This makes material travel across the sieves according to its particle size and sieve/ screen size.
Separator subclasses as per SUPAC: Vibratory/ Shaker and Centrifugal.
101. What is the gyratory motion of a vibro sifter and how does it get generated?
In the vibro sifter gyratory motion is obtained from a specially designed gyro motor, which is fitted underneath the vibrating assembly. A specially designed rugged spring completely isolates this assembly from the base with the help of Gyro-motor.

The motor is fitted with eccentric weights present at its top as well as base to create centrifugal force. This whole assembly is covered by an SS plate.
102. What are other names of vibro sifters?
The vibro sifter machine is also known as:
Vibro Screen
Vibrating Screen
Lab Vibro Sifter
Pharmaceutical Sifter
Vibro Sieve
Vibro Sifter Machine
Powder Sieving Machine
103. What are the salient features or use of vibro sifter?
• Gradation and separation of dry powder, granules, semi solids and liquids
• Main functioning mechanical arrangement is suspended on a spring to prevent vibration on the floor
• Easy assembling and disabling
• Easy to clean
104. What are different parts of vibro sifter?
• Dust cover
• Clamp for assembling of different parts
• Inlet
• Sieve/ screen
• Vibrating motor for Gyratory motion with eccentrically arranged top and bottom hammer
• Caster wheel
• Discharge port
• Spring
105. How to discharge the static electricity which may get generated over the vibro sifter sieve when silicone bonded sieve is used?
Following are the solutions to discharge the static electricity which may get generated over the vibro sifter sieve when silicone bonded screen is used:
i. Using antistatic screen
ii. Use C clamp over the silicone bonded sieve which makes the entire assembly conductive and to provide earthing to the body of the equipment.
106. What is a solution preparation vessel?
The solution preparation vessel is a closed tank used to prepare solution at room temperature or elevated temperature by using electrical heating or steam heating. To mix the solution homogeneously, it consists of stirring to create a vortex inside the solution.
107. What is the use of solution preparation vessels in the pharmaceutical industry?
The solution preparation vessel is used to prepare various types of solutions such as binder solution, coating solution, heating of water, liquid preparations such as syrup, suspension, solutions, etc. The vessel is used to ensure a homogeneous mix.
108. Explain parts of solution preparation vessel and Material of Construction (MOC)?
• Tank – SS316 or SS316L
• Stirrer – SS316 or SS316L
• Thermal Insulation
• Motor
• Variable Frequency Drive
• Control Panel
• Pressure gauge for jacket
• Temperature sensor – Product contact part SS316 or SS316 L
• Discharge valve (Zero dead leg and sanitary type)(Note: Ball valve is not acceptable) – SS316 or SS316L
• Caster wheel for movement (Need based) – Polyurethane
109. What is vortex and how is it helpful for solution preparation?
A circular, spiral, or helical motion in a fluid. A vortex often forms around areas of low pressure and attracts the fluid (and the objects moving within it) toward its center.
Velocity in the vortex is maximum next to the axis and inversely decreases with the radius.
Vortex enhances the desired solubility of pharmaceutical binder solute into the various solvents.

110. What should be characteristics of the solution preparation vessel?
• Zero dead leg and sanitary design components
• Stationary or mobile use
• Capable to handle wide temperature range (design as per requirement)
• The Internal surfaces of our preparation vessels are flush ground and mirror polished to <0.3 Microns Ra and electro-polished
• Easy to clean
• Adjustable speed of stirrer
• Adjustable temperature
• 21 CFR Part 11 compliant and EU Annex 11 Compliant
• Low maintenance and long service
111. What are the different particle size reduction mechanisms?
| Particle size reduction mechanisms | Description |
| Impact | Particle size reduction by applying an instantaneous force perpendicular to the particle/ agglomerate surface. The force can result from particle-to-particle or particle-to-mill surface collision. |
| Attrition | Particle size reduction by applying a force in a direction parallel to the particle surface. |
| Compression | Particle size reduction by applying a force slowly (as compared to Impact) to the particle surface in a direction toward the center of the particle. |
| Cutting | Particle size reduction by applying a shearing force to a material. |
112. What are the types of Equipment used for Particle Size Reduction/ Separation. Explain its Operating Principles.
| Types of Equipment | Operating Principles |
| Fluid Energy Milling | Particles are reduced in size as a result of high-speed particle-to-particle impact and/or attrition; also known as micronizing. |
| Impact Milling | Particles are reduced in size by high-speed mechanical impact or impact with other particles; also known as milling, pulverizing, or comminuting. |
| Cutting | Particles are reduced in size by mechanical shearing. |
| Compression Milling | Particles are reduced in size by compression stress and shear between two surfaces. |
| Screening | Particles are reduced in size by mechanically induced attrition through a screen. This process commonly is referred to as milling or deagglomeration. |
| Tumble Milling | Particles are reduced in size by attrition utilizing grinding media. |
| Separating | Particles are segregated based upon particle size alone and without any significant particle size reduction. This process commonly is referred to as screening or bolting. |
113. Explain Equipment Classifications for Particle Size Reduction/ Separation. Give examples of Equipment for each classification.
| Equipment type | Sub classification description | Sub classification |
| Fluid Energy Mills | Fluid energy mill subclasses have no moving parts and primarily are distinguished from one another by the configuration and/ or shape of their chambers, nozzles, and classifiers. | Tangential Jet Loop/ Oval Opposed Jet Opposed Jet with Dynamic Classifier Fluidized Bed Fixed Target Moving Target High Pressure Homogenizer E.g. Jet mill |
| Impact Mills | Impact mill subclasses primarily are distinguished from one another by the configuration of the grinding heads, chamber grinding liners (if any), and classifiers. | Hammer Air Swept Hammer Conventional Pin/ Disc Cage E.g. Hammer millPin mill Impact millCage millDisk millBall mill |
| Cutting Mills | Although cutting mills may differ from one another in whether the knives are movable or fixed and in the classifier configuration, no cutting mill subclasses have been identified. | No sub class E.g. MultimillComill |
| Compression Mills | Although compression mills may differ from one another in whether one or both surfaces are moving, no compression mill subclasses have been identified. | No sub class E.g. Roller mill |
| Screening Mills | Screening mill subclasses primarily are distinguished from one another by the rotating element. | Rotating Impeller Rotating Screen Oscillating Bar E.g. MultimillComill |
| Tumbling Mills | Tumbling mill subclasses primarily are distinguished from one another by the grinding media used and by whether the mill is vibrated. | Ball Media Rod Media Vibrating E.g. Ball millTubular Rod Mills |
| Separators | Separator subclasses primarily are distinguished from one another by the mechanical means used to induce particle movement. | Vibratory/ Shaker Centrifugal E.g. Vibro sifterUltra Centrifugal Mill |
Source: fda.gov
114. What is Blending and Mixing?
Blending and Mixing is the process of reorientation of particles relative to one another in order to achieve uniformity.
115. What are the Operating Principles of different equipment for Blending and Mixing?
| Types of Equipment | Operating Principles |
| Diffusion Blending (Tumble blending) | Particles are reoriented in relation to one another when they are placed in random motion and interparticular friction is reduced as the result of bed expansion (usually within a rotating container). |
| Convection Mixing (also known as paddle or plow mixing) | Particles are reoriented in relation to one another as a result of mechanical movement. |
| Pneumatic Mixing | Particles are reoriented in relation to one another as a result of the expansion of a powder bed by gas. |
116. Equipment Classifications used for Blending and Mixing
| Equipment type | Sub classification description | Sub classification |
| Diffusion Mixers (Tumble) | Diffusion mixer subclasses primarily are distinguished by geometric shape and the positioning of the axis of rotation. | • V-blenders• Double Cone Blenders• Slant Cone Blenders• Cube Blenders• Bin Blenders• Horizontal/ Vertical/ Drum Blenders• Static Continuous Blenders• Dynamic Continuous Blenders |
| Convection Mixers | Convection blender subclasses primarily are distinguished by vessel shape and impeller geometry. | • Ribbon Blenders• Orbiting Screw Blenders• Planetary Blenders• Forberg Blenders• Horizontal Double Arm Blenders• Horizontal High Intensity Mixers• Vertical High Intensity Mixers• Diffusion Mixers (Tumble) with Intensifier/Agitator |
| Pneumatic Mixers | Although pneumatic mixers may differ from one another in vessel geometry, air nozzle type, and air nozzle configuration, no pneumatic mixer subclasses have been identified. | No sub lass |
Source: fda.gov
117. What is Granulation
Granulation is the process of creating granules. The powder morphology is modified through the use of either a liquid that causes particles to bind through capillary forces or dry compaction forces.
The process will result in one or more of the following powder properties:
Enhanced flow;
Increased compressibility;
Densification;
Alteration of physical appearance to more spherical, uniform, or larger particles;
And/or enhanced hydrophilic surface properties.
118. What are the Operating Principles of different Granulation?
| Types of Granulation | Operating Principles |
| Dry Granulation | Dry powder densification and/or agglomeration by direct physical compaction. |
| Wet High-Shear Granulation | Powder densification and/or agglomeration by the incorporation of a granulation fluid into the powder with high-power-per-unit mass, through rotating high-shear forces. |
| Wet Low-Shear Granulation | Powder densification and/or agglomeration by the incorporation of a granulation fluid into the powder with low-power-per-unit mass, through rotating low-shear forces. |
| Low-Shear Tumble Granulation | Powder densification and/or agglomeration by the incorporation of a granulation fluid into the powder with low-power-per-unit mass, through rotation of the container vessel and/or intensifier bar. |
| Extrusion Granulation | Plasticization of solids or wetted mass of solids and granulation fluid with linear shear through a sized orifice using a pressure gradient. |
| Rotary Granulation | Spheronization, agglomeration, and/or densification of a wetted or non-wetted powder or extruded material. This is accomplished by centrifugal or rotational forces from a central rotating disk, rotating walls, or both. The process may include the incorporation and/or drying of a granulation fluid. |
| Fluid Bed Granulation | Powder densification and/or agglomeration with little or no shear by direct granulation fluid atomization and impingement on solids, while suspended by a controlled gas stream, with simultaneous drying. |
| Spray Dry Granulation | A pumpable granulating liquid containing solids (in solution or suspension) is atomized in a drying chamber and rapidly dried by a controlled gas stream, producing a dry powder. |
| Hot-melt Granulation | An agglomeration process that utilizes a molten liquid as a binder(s) or granulation matrix in which the active pharmaceutical ingredient (API) is mixed and then cooled down followed by milling into powder. This is usually accomplished in a temperature controlled jacketed high shear granulating tank or using a heated nozzle that sprays the molten binders(s) onto the fluidizing bed of the API and other inactive ingredients. |
| Melt Extrusion | A process that involves melting and mixing API and an excipient (generally a polymer) using low or high shear kneading screws followed by cooling and then milling into granules. Thermal energy for melting is usually supplied by the electric/water heater placed on the barrel. Materials are either premixed or fed into an extruder separately. Melt extruder subclasses primarily are distinguished by the configuration of the screw. • Single screw extruder • Twin screw extruder |
119. Equipment Classifications used for granulation.
| Equipment type | Sub classification description | Sub classification |
| Dry Granulator | Dry granulator subclasses primarily are distinguished by the densification force application mechanism. | • Slugging • Roller Compaction |
| Wet High-Shear Granulator | Wet high-shear granulator subclasses primarily are distinguished by the geometric positioning of the primary impellers; impellers can be top, bottom, or side driven. | • Vertical (Top or Bottom Driven) • Horizontal (Side Driven) |
| Wet Low-Shear Granulator | Wet low-shear granulator subclasses primarily are distinguished by the geometry and design of the shear inducing components; shear can be induced by rotating impeller, reciprocal kneading action, or convection screw action. | • Planetary • Kneading • Screw |
| Low-Shear Tumble Granulator | Although low-shear tumble granulators may differ from one another in vessel geometry and type of dispersion or intensifier bar, no low-shear tumble granulator subclasses have been identified. | • Slant cone • Double cone • V-blender |
| Extrusion Granulator | Extrusion granulator subclasses primarily are distinguished by the orientation of extrusion surfaces and driving pressure production mechanism. | • Radial or Basket • Axial • Ram • Roller, Gear, or Pelletizer |
| Rotary Granulator | Rotary granulator subclasses primarily are distinguished by their structural architecture. They have either open top architecture, such as a vertical centrifugal spheronizer, or closed top architecture, such as a closed top fluid bed dryer. | • Open • Closed |
| Fluid Bed Granulator | Although fluid bed granulators may differ from one another in geometry, operating pressures, and other conditions, no fluid bed granulator subclasses have been identified. | No subclass |
| Spray Dry Granulator | Although spray dry granulators may differ from one another in geometry, operating pressures, and other conditions, no spray dry granulator subclasses have been identified. | No subclass |
| Hot-melt Granulator | Although, hot-melt granulator may differ from one another in primarily melting the 455 inactive ingredient (particularly the binder or other polymeric matrices), no 456 subclasses have been identified at this time. | No subclass |
120. What is an integrated unit as per SUPAC?
When a single piece of equipment is capable of performing multiple discrete unit operations (i.e. mixing, granulating, drying), the unit was evaluated solely for its ability to granulate. If multifunctional units were incapable of discrete steps (fluid bed granulator/ drier), the unit was evaluated as an integrated unit.
Source: fda.gov
121. What is Drying?
The removal of a liquid from a solid by evaporation.
122. What are the Operating Principles of different equipment for Drying?
| Types of Drying principle | Details of Principles |
| Direct Heating, Static Solids Bed | Heat transfer is accomplished by direct contact between the wet solids and hot gases. The vaporized liquid is carried away by the drying gases. There is no relative motion among solid particles. The solids bed exists as a dense bed, with the particles resting upon one another. |
| Direct Heating, Moving Solids Bed | Heat transfer is accomplished by direct contact between the wet solids and hot gases. The vaporized liquid is carried away by the drying gases. Solids motion is achieved by either mechanical agitation or gravity force, which slightly expands the bed enough to flow one particle over another. |
| Direct Heating, Fluidized Solids Bed | Heat transfer is accomplished by direct contact between the wet solids and hot gases. The vaporized liquid is carried away by the drying gases. The solids are in an expanded condition, with the particles supported by drag forces caused by the gas phase. The solids and gases intermix and behave like a boiling liquid. This process commonly is referred to as fluid bed drying. |
| Direct Heating, Dilute Solids Bed, Spray Drying | Heat transfer is accomplished by direct contact between a highly dispersed liquid and hot gases. The feed liquid may be a solution, slurry, emulsion, gel or paste, provided it is pumpable and capable of being atomized. The fluid is dispersed as fine droplets into a moving stream of hot gases, where they evaporate rapidly before reaching the wall of the drying chamber. The vaporized liquid is carried away by the drying gases. The solids are fully expanded and so widely separated that they exert essentially no influence on one another. |
| Direct Heating, Dilute Solids Bed, Flash Drying | Heat transfer is accomplished by direct contact between wet solids and hot gases. The solid mass is suspended in a finely divided state in a high-velocity and high-temperature gas stream. The vaporized liquid is carried away by the drying gases. |
| Indirect Conduction, Moving Solids Bed | Heat transfer to the wet solid is through a retaining wall. The vaporized liquid is removed independently from the heating medium. Solids motion is achieved by either mechanical agitation or gravity force, which slightly expands the bed enough to flow one particle over another. |
| Indirect Conduction, Static Solids Bed | Heat transfer to the wet solid is through a retaining wall. The vaporized liquid is removed independently from the heating medium. There is no relative motion among solid particles. The solid bed exists as a dense bed, with the particles resting upon one another. |
| Indirect Conduction, Lyophilization | Drying in which the water vapor sublimes from the product after freezing. |
| Gas Stripping | Heat transfer is a combination of direct and indirect heating. The solid motion is achieved by agitation and the bed is partially fluidized. |
| Indirect Radiant, Moving Solids Bed | Heat transfer is accomplished with varying wavelengths of energy. Vaporized liquid is removed independently from the solid bed. The solid’s motion is achieved by mechanical agitation, which slightly expands the bed enough to flow one particle over one another. This process commonly is referred to as microwave drying. |
123. Equipment Classifications used for drying.
| Equipment type | Sub classification description | Sub classification |
| Direct Heating, Static Solids Bed | Static solids bed subclasses primarily are distinguished by the method of moving the solids into the dryer. | • Tray and Truck • Belt |
| Direct Heating, Moving Solids Bed | Moving solids bed subclasses primarily are distinguished by the method or technology for moving the solid bed. | • Rotating Tray • Horizontal Vibrating Conveyor |
| Direct Heating, Fluidized Solids Bed (Fluid Bed Dryer) | Although fluid bed dryers may differ from one another in geometry, operating pressures, and other conditions, no fluidized solids bed dryer subclasses have been identified. | No subclass |
| Direct Heating, Dilute Solids Bed, Spray Dryer | Although spray dryers may differ from one another in geometry, operating pressures, and other conditions, no spray dryer subclasses have been identified. | No subclass |
| Direct Heating, Dilute Solids Bed, Flash Dryer | Although flash dryers may differ from one another in geometry, operating pressures, and other conditions, no flash dryer subclasses have been identified. | No subclass |
| Indirect Conduction Heating, Moving Solids Bed | Moving solids bed subclasses primarily are distinguished by the method or technology for moving the solids bed. | • Paddle • Rotary (Tumble) • Agitation |
| Indirect Conduction Heating, Static Solids Beds | No indirect heating, static solids bed shelf dryer subclasses have been identified. | No subclass |
| Indirect Conduction, Lyophilization | No lyophilizer subclasses have been identified. | No subclass |
| Gas Stripping | Although gas stripping dryers may differ from one another in geometry, shape of agitator, and how fluidizing gas is moved through the bed, no gas stripping dryer subclasses have been identified. | No subclass |
| Indirect Radiant Heating, Moving Solids Bed (Microwave Dryer) | Although microwave dryers may differ from one another in vessel geometry and the way microwaves are directed into the solids, no indirect radiant heating, moving solids bed dryer subclasses have been identified. | No subclass |
Source: fda.gov
124. What is Unit Dosing?
The division of a powder blend into uniform single portions for delivery to patients.
125. What are the Operating Principles of different equipment for Unit Dosing?
| Types of Unit Dosing principle | Details of Principles |
| Tabletting | The division of a powder blend in which compression force is applied to form a single unit dose. |
| Encapsulating | The division of material into a hard gelatin capsule. Encapsulators should all have the following operating principles in common: rectification (orientation of the hard gelatin capsules), separation of capsule caps from bodies, dosing of fill material/formulation, rejoining of caps and bodies, and ejection of filled capsules. |
| Powder Filling | The division of a powder blend into a container closure system. |
126. Equipment Classifications used for Unit Dosing.
| Equipment type | Sub classification description | Sub classification |
| Tablet Press | Tablet press subclasses primarily are distinguished from one another by the method that the powder blend is delivered to the die cavity. Tablet presses can deliver powders without mechanical assistance (gravity), with mechanical assistance (power assisted), by rotational forces (centrifugal), and in two different locations where a tablet core is formed and subsequently an outer layer of coating material is applied (compression coating). | • Gravity• Power Assisted• Centrifugal• Compression Coating |
| Tablet Press | Tablet press subclasses are also distinguished from one another for some special types of tablets where more than one hopper and precise powder feeding mechanism might be necessary. | • Multi-tablet press for micro/mini tablet • Multi-layer tablet press (bi-layer, tri-layer) |
| Encapsulator | Encapsulator subclasses primarily are distinguished from one another by the method that is used for introducing material into the capsule. Encapsulators can deliver materials with a rotating auger, vacuum, vibration of perforated plate, tamping into a bored disk (dosing disk), or cylindrical tubes fitted with pistons (dosator). | • Auger• Vacuum• Vibratory• Dosing Disk• Dosator |
| Powder Filler | Subclasses of powder fillers primarily are distinguished by the method used to deliver the predetermined amount for container fill. | • Vacuum• Auger |
Source: fda.gov
127. What is Coating?
The uniform deposition of a layer of material on or around a solid dosage form.
128. Why is the coating done?
Coating is done for the following reasons:
a. Protect the drug from its surrounding environment (air, moisture, and light), with a view to improving stability.
b. Mask unpleasant taste, odor, or color of the drug.
c. Increase the ease of ingesting the product for the patient.
d. Impart a characteristic appearance to the tablets, which facilitates product identification and aids patient compliance.
e. Provide physical protection to facilitate handling. This includes minimizing dust generation in subsequent unit operations.
f. Reduce the risk of interaction between incompatible components. This would be achieved by coating one or more of the offending ingredients.
g. Modify the release of the drug from the dosage form. This includes delaying, extending, and sustaining drug substance release.
h. Modify the dosage form by depositing the API or drug substance on or around a core tablet, which could be a placebo core tablet or a tablet containing another drug or a fractional quantity of the same drug.
129. What are the major techniques used for coating?
The coating material deposition typically is accomplished through one of six major techniques:
a. Sugar Coating – Deposition of coating material onto the substrate from aqueous solution/suspension of coatings, based predominantly upon sucrose as a raw material.
b. Film Coating – The deposition of polymeric film onto the solid dosage form.
c. Core Enrobing – The gelatin coating of gravity or force fed pre- formed tablets or caplets.
d. Microencapsulation – The deposition of a coating material onto a particle, pellet, granule, or bead core. The substrate in this application ranges in size from submicron to several millimeters. It is this size range that differentiates it from the standard coating described in 1 and 2 above.
e. Compression Coating (also addressed in the Unit Dosing section) – A coating process where a dry coatings blend is applied on a previously compressed core tablet using a tablet compression machine.11 Therefore, this process is also known as a dry coating process that does not involve any water or any other solvent in the coating process.
f. Active/ API coating – Deposition of active pharmaceutical ingredient (API or drug substance) on or around a core tablet utilizing any of the above five coating techniques.
Source: fda.gov
130. What are the Operating Principles of different equipment for coating?
| Types of coating | Details of Principles |
| Pan Coating | The uniform deposition of coating material onto the surface of a solid dosage form, or component thereof, while being translated via a rotating vessel. |
| Gas Suspension | The application of a coating material onto a solid dosage form, or component thereof, while being entrained in a process gas stream. Alternatively, this may be accomplished simultaneously by spraying the coating material and substrate into a process gas stream. |
| Vacuum Film Coating | This technique uses a jacketed pan equipped with a baffle system. Tablets are placed into the sealed pan, an inert gas (i.e., nitrogen) is used to displace the air and then a vacuum is drawn. |
| Dip Coating | Coating is applied to the substrate by dipping it into the coating material. Drying is accomplished using pan coating equipment. |
| Electrostatic Coating | A strong electrostatic charge is applied to the surface of the substrate. The coating material containing oppositely charged ionic species is sprayed onto the substrate. |
| Compression Coating | The division of a powder blend in which compression force is applied to form a single unit dose. |
131. Equipment Classifications used for coating.
| Equipment type | Sub classification description | Sub classification |
| Pan Coating | Pan coating subclasses primarily are distinguished by the pan configuration, the pan perforations, and/or the perforated device used to introduce process air for drying purposes. Perforated coating systems include both batch and continuous coating processes. | • Non-perforated (conventional) Coating System • Perforated Coating System |
| Gas Suspension | Gas suspension subclasses primarily are distinguished by the method by which the coating is applied to the substrate. | • Fluidized Bed with bottom spray mechanism • Fluidized Bed with tangential spray mechanism • Fluidized Bed with top spray mechanism • Fluidized Bed with Wurster column • Spray Congealing/Drying |
| Vacuum Film Coating | Although there may be differences in the jacketed pan, baffle system, or vacuum source, no vacuum film coating subclasses have been identified. | No sub class |
| Dip Coating | Because of the custom design associated with this class of coating, no dip 1013 coating subclasses or examples have been identified. | No sub class |
| Electrostatic Coating | Because of the custom design associated with this class of coating, no 1018 electrostatic coating subclasses or examples have been identified. | No sub class |
| Compression Coating | Refer tablet press principle Unit Dosing section of previous post. | No sub class |
Source: fda.gov
132. What is Printing?
The marking of a capsule or tablet surface for the purpose of product identification.
Printing may be accomplished by either the application of a contrasting colored polymer (ink) onto the surface of a capsule or tablet, or by the use of laser etching. The method of application, provided the ink formulation is not altered, is of no consequence to the physical-chemical properties of the product.
133. What is Drilling?
The drilling or ablating of a hole or holes through the polymeric film coating shell on the surfaces of a solid oral dosage form using a laser.
The polymeric film shell is not soluble in vivo. The hole or holes allow for the modified release of the drug from the core of the dosage form.
134. What are the Operating Principles of different equipment for Printing and Drilling?
| Types of Printing/ Drilling | Details of Principles |
| Ink-Based Printing | The application of contrasting colored polymer (ink) onto the surface of a tablet or capsule. |
| Laser Etching | The application of identifying markings onto the surface of a tablet or capsule using laser-based technology. |
| Drilling | A drilling system typically is a custom built unit consisting of a material handling system to orient and hold the solid dosage form, a laser (or lasers), and optics (lenses, mirrors, deflectors, etc.) to ablate the hole or holes, and controls. The drilling unit may include debris extraction and inspection systems as well. The sorting, orienting, and holding equipment commonly is provided by dosage form printing equipment manufacturers, and is considered ancillary in this use. |
135. Equipment Classifications used for Printing and Drilling.
| Equipment type | Sub classification description | Sub classification |
| Ink-Based Printing | Ink-based printing subclasses primarily are distinguished by the method by which the marking is applied to a capsule or tablet surface. | • Offset • Ink Jet |
| Laser Etching (Printing) | Although laser etching systems may differ from one another, no laser etching subclasses have been identified. | No sub class |
| Drilling | The method of producing the laser pulse that ablates the hole(s) is of no consequence to the physical-chemical properties of the product. Therefore, no dosage form drilling equipment subclasses have been identified. | No sub class |
Source: fda.gov
136. What is excipient?
The word excipient originates from the Latin excipere, which means to receive; hence, the excipient receives the active substance.
As per European Pharmacopoeia (Ph. Eur.) “An excipient is any component, other than the active substance(s), present in a medicinal product or used in the manufacture of the product. The intended function of an excipient is to act as the carrier (vehicle or basis) or as a component of the carrier of the active substance(s) and, in so doing, to contribute to product attributes such as stability, biopharmaceutical profile, appearance and patient acceptability and to the ease with which the product can be manufactured. Usually, more than one excipient is used in the formulation of a medicinal product.”
147. What are the typical functions of excipients?
Excipients play a role in the formulation as Diluent, Binder, Disintegrant, Glidant, Lubricant, Coatings agent, or Coloring agent. It can further classified depending on its functions in the dosage form, which includes:
(i) Modulating solubility and bioavailability of the drug,
(ii) Enhancing stability of the drug in its dosage forms,
(iii) Maintaining a required polymorphic form,
(iv) Maintaining pH and osmolarity of liquid products,
(v)Antioxidants,
(vi) Preventing aggregation or dissociation agent,
(vii) Modulating the immunogenic response of drug,
(viii) Suspending agent,
(ix) Emulsifier,
(x) Aerosol propellants,
(xi) Base or tablet diluent
138. What is role of Diluents as an excipient?
Diluents are the material which provides bulk to the formulation and enable accurate dosing of potent ingredients.
139. Give a few examples of Diluents as an excipient?
Microcrystalline cellulose, Lactose, dextrin, glucose, sucrose, sorbitol, silicates, calcium and magnesium salts, sodium or potassium chloride, starch, lactose
140. What is the role of Binder as an excipient?
Binders, compression aids, granulating agents are the materials which bind the ingredients together giving form and mechanical strength and form granules for tablet, capsule or relevant formualtions
141. Give a few examples of Binder as an excipient?
Polyvinyl pyrrolidone, starch, gelatin, cellulose derivatives, sugars, sugar alcohols and cellulose derivatives
142. What is the role of Disintegrant as an excipient?
Disintegrant aid in dispersion of the tablet in the gastrointestinal tract, releasing the active ingredient and increasing the surface area for dissolution.
143. Give a few examples of Disintegrant as an excipient?
Starch, sodium starch glycollate, cross – linked polyvinyl pyrrolidone super disintegrants, cellulose derivatives and alginates, and crospovidone.
144. What is role of Glidant as an excipient?
Glidant improves the flow of powders during tablet manufacturing by reducing friction and adhesion between particles. It also used as anti-caking agents.
145. Give a few examples of Glidant as an excipient?
Colloidal anhydrous silicon and other silica compounds, talc, magnesium stearate
146. What is the role of Lubricant as an excipient?
Lubricants are used to prevent sticking of granules from die and punch wall during compression.
147. Give a few examples of Lubricant as an excipient?
Stearic acid and its salts (e.g. magnesium stearate), polyethylene glycol, sodium chloride
148. What is the role of coating agents as an excipient?
Coating agents have various functions such as, protect tablets from the environment (air, light and moisture), increase the mechanical strength, mask taste and smell, aid swallowing, assist in product identification.
It also can be used to modify release of the active ingredient.
149. Give a few examples of coating agents as an excipient?
Sugar, cellulose acetate phthalate etc.
150. What is the role of Colouring agent as an excipient?
Coloring agents improves acceptability to patients, aids identification and prevents counterfeiting. It also increases the stability of light-sensitive drugs.
151. Give a few examples of Colouring agents as an excipient?
Iron oxide, natural pigments, and other synthetic dyes.
152. Explain excipient classification based on objective of addition in dosage form.
| Type of excipient | Objective in dosage form |
| Improve organoleptic property | Color Flavor Sweetener Masking of unpleasant test |
| Stabilizers | Preservative Antioxidant Emulsifier Suspending agent Isotonicity agent Maintaining pH and osmolarity |
| Dosage accuracy | Diluent Filler Bulking agent |
| Process aids | Binder Glidant Lubricant Anti-adherent |
| Drug release | Disintigrant Permiability enhancer Release rate limiting agent |
153. What is immediate – release dosage form?
This is the dosage form intended to release the drug immediately after administration.
154. What is Delayed – release dosage form?
This is the dosage form where drug is not released until a physical event has occurred, e.g., change in pH etc.
155. What is sustained – release dosage form?
This is the dosage form where drug is released slowly over extended time.
156. What is a soluble tablet?
Soluble tablets are those which dissolve in water before administration.
157. What is a Dispersible tablet?
Tablet is added to water to form a suspension to administer.
158. What is an Effervescent tablet?
Tablet which is added into water. Tablet releases carbon dioxide to form an effervescent solution.
159. What is a Chewable tablet?
To administer the dose, the tablet is chewed and swallowed.
160. What is Chewable gum?
The formulation is chewed and removed from the mouth after a directed time as per the label claim.
161. What are Buccal and sublingual tablets?
The tablet which is placed in the oral cavity for local or systemic action is called Buccal and sublingual.
162. What is an Orally disintegrating tablet?
The tablet which gets dissolved or disintegrated in the mouth without the need for water is called an orally disintegrating tablet.
163. What is Lozenge?
Lozenge is a slowly dissolving tablet designed to be sucked in the mouth.
164. What is Pastille?
Pastille is the tablet consisting of gelatin and glycerine that facilitate dissolving tablets slowly in the mouth.
165. What is a Hard gelatin capsule?
Hard gelatin capsule is a two piece capsule shell that is filled with powder, granulate, tablets, semisolid or liquid in it.
166. What is Soft gelatin capsule (softgel)?
Soft gelatin capsule is a one piece capsule that contains a liquid or semisolid filled in it.
References:
1. WHO GMP Guidelines: Guide to Master Formulae, WHO/FWC/IVB/QSS/VQR, 2011
EU and PIC GMP guidelines: EudraLex Volume 4, Chapter 4: Documentation
PIC/S guidelines: Chapter 4: Documentation
Health Canada GMP guidelines: Good manufacturing practices guide for drug products (GUI-0001), Manufacturing control, C.02.011
U.S. FDA: CFR 21, Chapter I, Subchapter F: Biologics, Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals; Subpart F–Production and Process Controls, Sec. 211.100 Written procedures; deviations; and Subpart J–Records and Reports; Sec. 211.186 Master production and control records
U.S. FDA: CFR 21, Chapter I, Subchapter F: Biologics; Subchapter C: Drugs General; Part 211 Current Good Manufacturing Practice for Finished Pharmaceuticals; Subpart J– Records and Reports; Sec. 211.188 Batch production and control records.
India: The drugs and cosmetics act, 1940 and The drugs and cosmetics rules, 1945, Schedule M, 12. Documentation and records]
2. PART 314 — APPLICATIONS FOR FDA APPROVAL TO MARKET A NEW DRUG, Subpart A – General Provisions Sec. 314.3 Definitions.
3. 21 CFR PART 210: CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING, PROCESSING, PACKING, OR HOLDING OF DRUGS; GENERAL Sec. 210.1 Status of current good manufacturing practice regulations.
5. Wet Granulation:
End-Point Determination and Scale-Up, By Michael Levin, Ph. D., Metropolitan Computing Corporation East Hanover, New Jersey, USA
6. Saudi Pharmaceutical Journal
Volume 20, Issue 1, January 2012, Pages 9-19, Saudi Pharmaceutical Journal, Review article, Upgrading wet granulation monitoring from hand squeeze test to mixing torque rheometry Author links open overlay panel Walid F. Sakr Mohamed A. Ibrahim Fars K. Alanazi Adel A. Sakr
7. natoli.com
8. pacifictools.in

125+ Pharmaceutical Interview Questions and Answers for Sterile Formulations
This page covers most of the interview questions and answers during a technical round in Production of Sterile Formulations. The interview questions cover questions from basic to advance level of technical aspects. These interview questions and answers will help to crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
The topics covered here are sterilization process, aseptic processing, media fill, area classification, and associated topics. In addition, the interview questions and answers cover various equipment used for the manufacturing process of sterile formulations.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning, and best of luck with your interview! Happy Learning.
1. What is sterilization:
A suitably designed, validated and controlled process that inactivates or removes viable microorganisms in a product until sterility is obtained.
2. What is Sterility:
Sterility is the absence of viable microorganisms, as defined by a sterility assurance level equal to or less than 10−6. The inactivation of microorganisms by physical or chemical means follows an exponential law; thus there is always a finite statistical probability that a micro-organism may survive the sterilizing process. For a given process, the probability of survival is determined by the number, types and resistance of the microorganisms present and by the environment in which the organisms exist during treatment.
3. What is Aseptic processing?
A process performed maintaining the sterility of a product that is assembled from components, each of which has been sterilised by steam, dry heat, ionizing radiation, gas or sterile filtration. This is achieved by using conditions and facilities designed to prevent microbiological contaminants.
4. Bioburden:
The total number of micro-organisms associated with a specific item prior to any sterilisation or bioburden reduction step.
5. Biological indicator:
Biological indicators are test systems containing viable microorganisms (usually spores of bacteria) that provide a defined challenge to verify the required effectiveness of a specified sterilisation process.
6. Colony Forming Unit (CFU):
A microbiological term that describes the formation of a single macroscopic colony after the introduction of one or more micro-organisms to microbiological growth media. One colony forming unit is expressed as 1 CFU.
7. Depyrogenation
A process used to destroy or remove pyrogens (e.g. endotoxins).
8. D-value (decimal reduction value)
The value of a parameter of sterilisation (duration or absorbed dose) required to reduce the number of viable organisms to 10 per cent of the original number. It is only of significance under precisely defined experimental conditions. D121 is the D-value of the relevant spores at 121° C.
9. F0 value
The F0 value of a saturated steam sterilization process is the lethality expressed in terms of the equivalent time in minutes at a temperature of 121 °C delivered by the process to the load in its container with reference to micro-organisms possessing a theoretical Z-value of 10.
10. Holding time
The time between two process steps.
11. Lethal (process)
A process that kills the microorganisms exponentially.
12. Overkill sterilization
A process with a lethality of F0BIO > 12 minutes. For example a process that provides at least a 12 log reduction of biological indicator microorganisms having a minimum D value of 1 minute.
13. Ph. Eur. sterilization reference conditions
The reference conditions for sterilisation specified in Ph. Eur. 5.1.1, i.e. terminal steam sterilization at ≥121 °C for 15 min, terminal dry heat sterilisation at ≥160 °C for ≥2 h or terminal ionising radiation of 25 kGy.
14. Post-aseptic processing terminal heat treatment
A terminal moist heat process employed after aseptic processing which has been demonstrated to provide a SAL ≤10-6, but where the requirements of steam sterilisation (for example, F0≥8 min) are not fulfilled.
15. SAL (Sterility Assurance Level)
The SAL for a given sterilisation process is expressed as the probability of micro-organisms surviving in a product item after exposure to the process. An SAL of 10-6, for example, denotes a probability of not more than 1 non-sterile item in 1 × 106 sterilised items of the final product.
16. TAMC (Total aerobic microbial count)
The total aerobic microbial count (TAMC) is considered to be equal to the number of CFU found using casein soya bean digest agar.
17. z-value
The z-value is the change in temperature required to alter the D-value by a factor of 10.
Reference for Q 1 to 17:: 6 March 2019 EMA/CHMP/CVMP/QWP/850374/2015, Committee for Medicinal Products for Human use (CHMP), Committee for Medicinal Products for Veterinary use (CVMP), Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container
18. Difference Between F0 and Fh Values
At this point of time, it’ll be better to differentiate the two i.e. F0 and Fh values.
| F0 Value | Fh Value |
| Used to evaluate the effectiveness of Steam Sterilization | Used to evaluate the effectiveness of Dry Heat Sterilization |
| The assumed z-value is 10°C for sterilization range of 100 to 130°C | The assumed z-value is 20°C for sterilization range of 160 to 200°C |
| Theoretical Requirement121.1°C @ 30 min. of sterile hold time | Theoretical Requirement170°C @ 32 min. of sterile hold time |
| Targetted to mitigate micro-organisms especially living endospores | Targetted to remove the bacterial endotoxins |
| Much more complicated because of steam quality requirements | Lethality of the microbes is less than that of F0 at the same temperature |
19. What are the sterile manufacturing area grades? What are the operations to be carried out in each area?
For the manufacture of sterile medicinal products 4 grades can be distinguished:
Grade A: The local zone for high risk operations, e.g. filling zone, stopper bowls, open ampoules and vials, making aseptic connections. Normally such conditions are provided by a laminar air flow work station. Laminar air flow systems should provide a homogeneous air speed in a range of 0.36–0.54 m/s (guidance value) at the working position in open clean room applications. The maintenance of laminarity should be demonstrated and validated. A uni-directional air flow and lower velocities may be used in closed isolators and glove boxes.
Grade B: For aseptic preparation and filling, this is the background environment for the grade A zone.
Grade C and D: Clean areas for carrying out less critical stages in the manufacture of sterile products.
20. Explain correlation between Area Grades, Area Class and ISO Classification “At Rest” and “In Operation”.
| Grade | ISO Class number (At rest) | Class (At rest) | ISO Class number (At rest) | Class (At rest) |
| A | 4.8 | 100 | 4.8 | 100 |
| B | 5 | 100 | 7 | 10,000 |
| C | 7 | 10,000 | 8 | 100,000 |
| D | 8 | 100,000 | Not defined | Not defined |
Note:
For Grade A the airborne particle classification is ISO 4.8 dictated by the limit for particles ≥ 5.0 μm.
For Grade B (at rest) the airborne particle classification is ISO 5 for both considered particle sizes.
For Grade C (at rest and in operation) the airborne particle classification is ISO 7 and ISO 8 respectively.
For Grade D (at rest) the airborne particle classification is ISO 8. (In operation no classification is defined).
Reference – Rules and Guidance for Pharmaceutical Manufacturers and Distributors (MHRA)
21. Provide example of operations to be carried out in the various grades for sterile manufacturing facility.
Examples of operations to be carried out in the various grades are as follows:
| Grade | Examples of operations for terminally sterilised products. |
| A | Filling of products, when unusually at risk. |
| C | Preparation of solutions, when unusually at risk. Filling of products. |
| D | Preparation of solutions and components for subsequent filling. |
| Grade | Examples of operations for aseptic preparations. |
| A | Aseptic preparation and filling. |
| C | Preparation of solutions to be filtered. |
| D | Handling of components after washing. |
Reference – Rules and Guidance for Pharmaceutical Manufacturers and Distributors (MHRA)
22. Explain clothing requirements for each grade of manufacturing are for sterile manufacturing facility.
Grade A/B: Headgear should totally enclose hair and, where relevant, beard and moustache; it should be tucked into the neck of the suit; a face mask should be worn to prevent the shedding of droplets. Appropriate sterilised, non-powdered rubber or plastic gloves and sterilised or disinfected footwear should be worn. Trouser-legs should be tucked inside the footwear and garment sleeves into the gloves. The protective clothing should shed virtually no fibres or particulate matter and retain particles shed by the body.
Outdoor clothing should not be brought into changing rooms leading to grade B and C rooms. For every worker in a grade A/B area, clean sterile (sterilised or adequately sanitised) protective garments should be provided at each work session. Gloves should be regularly disinfected during operations. Masks and gloves should be changed at least for every working session.
Grade C. Hair and where relevant beard and moustache should be covered. A single or two-piece trouser suit, gathered at the wrists and with high neck and appropriate shoes or overshoes should be worn. They should shed virtually no fibres or particulate matter.
Grade D. Hair and, where relevant, beard should be covered. A general protective suit and appropriate shoes or overshoes should be worn. Appropriate measures should be taken to avoid any contamination coming from outside the clean area.
Reference – Rules and Guidance for Pharmaceutical Manufacturers and Distributors (MHRA)
23. Explain design consideration in sterile manufacturing area for contamination prevention and clean area separation
- Airflow direction shall be from areas of higher cleanliness to adjacent less clean areas.
- Higher air cleanliness shall have a substantial positive pressure differential relative to adjacent rooms of lower air cleanliness.
- Positive pressure differential of at least 10-15 Pascals (Pa) should be maintained between adjacent rooms of differing classification (with doors closed).
- When doors are open, outward airflow should be sufficient to minimize ingress of contamination, and it is critical that the time a door can remain ajar be strictly controlled.
- When unclassified room adjacent to the aseptic processing room, a substantial overpressure (e.g., at least 12.5 Pa) from the aseptic processing room should be maintained at all times to prevent contamination.
- Continuous monitoring of pressure differentials between cleanrooms with frequently recorded.
24. What is the air changes requirement for Class 100,000 (ISO 8)?
For Class 100,000 (ISO 8) supporting rooms, airflow sufficient to achieve at least 20 air changes per hour is typically acceptable.
Significantly higher air change rates are normally needed for Class 10,000 and Class 100 areas.
25. What is difference between filter leak testing and efficiency testing?
An efficiency test is a general test used to determine the rating of the filter. An intact HEPA filter should be capable of retaining at least 99.97 percent of particulates greater than 0.3 μm in diameter.
The purpose of leak test is to detect leaks from the filter media, filter frame, or seal.
26. What is the limit of HEPA filter leak test?
While performing leak test, a single probe reading equivalent to 0.01 percent of the upstream challenge would be considered as indicative of a significant leak.
27. Why measurement of velocity is important in the aseptic area? What should be measurement location and distance from filter face?
HEPA filter leak testing alone is insufficient to monitor filter performance. It is important to conduct periodic monitoring of filter attributes such as uniformity of velocity across the filter (and relative to adjacent filters). Variations in velocity can cause turbulence that increases the possibility of contamination. Velocities of unidirectional air should be measured 6 inches from the filter face and at a defined distance proximal to the work surface for HEPA filters in the critical area. Velocity monitoring at suitable intervals can provide useful data on the critical area in which aseptic processing is performed. The measurements should correlate to the velocity range established at the time of in situ air pattern analysis studies.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
28. Drains are preferably not allowed in which area classification zones?
Drains are considered inappropriate for classified areas of the aseptic processing facility other than Class 100,000 (ISO 8) areas.
29. Types of training required to be conducted before an individual is permitted to enter the aseptic manufacturing area?
Fundamental training topics should include aseptic technique, cleanroom behavior, microbiology, hygiene, gowning, patient safety hazards posed by a non-sterile drug product, and the specific written procedures covering aseptic manufacturing area operations.
30. What measures required to be followed by personnel in aseptic area to maintain sterility of sterile items and surafaces?
- Measures required to be followed by personnel in aseptic area are:
- Contact sterile materials only with sterile instruments.
- After initial gowning, sterile gloves should be regularly sanitized or changed, as appropriate, to minimize the risk of contamination.
- Personnel should not directly contact sterile products, containers, closures, or critical surfaces with any part of their gown or gloves.
- Move slowly and deliberately.
- Keep the entire body out of the path of unidirectional airflow.
- Approach a necessary manipulation in a manner that does not compromise sterility of the product.
- Maintain Proper Gown Control – Prior to and throughout aseptic operations, an operator should not engage in any activity that poses an unreasonable contamination risk to the gown.
31. What is the impact of rapid movements in Aspetic area?
Rapid movements can create unacceptable turbulence in a critical area. Such movements disrupt the unidirectional airflow, presenting a challenge beyond intended cleanroom design and control parameters. The principle of slow, careful movement should be followed throughout the cleanroom.
32. What is the potential impact if body parts interrupt the path of unidirectional airflow?
Disruption of the path of unidirectional flow air in the critical area can pose a risk to product sterility because, the purpose of unidirectional airflow design is to protect sterile equipment surfaces, container-closures, and product.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
33. Explain the type of gowning required for aseptic area?
- The gown should provide a barrier between the body and exposed sterilized materials and prevent contamination from particles generated by, and microorganisms shed from, the body.
- Gowns should be sterile and non-shedding, and cover the skin and hair (face-masks, hoods, beard/ moustache covers, protective goggles, and elastic gloves are examples of common elements of gowns).
- An adequate barrier should be created by the overlapping of gown components (e.g., gloves overlapping sleeves).
- If an element of a gown is found to be torn or defective, it should be changed immediately. Gloves should be sanitized frequently.
34. What is the recommended gowning requalification frequency for aseptic area?
Annual requalification is normally sufficient for those automated operations where personnel involvement is minimized and monitoring data indicate environmental control.
For any aseptic processing operation, if adverse conditions occur, additional or more frequent requalification could be indicated.
35. Sanitizing gloves just prior to sampling is acceptable or not acceptable?
Sanitizing gloves just prior to sampling is inappropriate because it can prevent recovery of microorganisms that were present during an aseptic manipulation.
36. What is the recommended solvent for rinse sampling for pre-sterilization preparation of glass containers? What is the acceptance criterion of final rinse water?
High purity water. Final rinse water should meet the specifications of WFI, USP.
37. What is a log reduction criterion for endotoxin after depyrogenation process?
Validation study data should demonstrate that the process reduces the endotoxin content by at least 99.9 percent (3 logs).
38. Is sterilizing-grade filters and moist heat sterilization effective in removing endotoxin?
Sterilizing-grade filters and moist heat sterilization have not been shown to be effective in removing endotoxin.
39. What is an effective way to inactivate endotoxins?
Endotoxin on equipment surfaces can be inactivated by high-temperature dry heat, or removed from equipment surfaces by cleaning procedures.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
40. What should be frequency of periodic media fill?
Semi-annual qualification should be conducted for each processing line to evaluate the state of control of the aseptic process.
41. What permutation and combination factors should be covered during the media fill?
Activities and interventions representative of each shift, and shift changeover, should be incorporated into the design of the semi-annual qualification program. E.g. Production shift should address its unique time-related and operational features.
42. What is the requirement of participation of personnel during media fill? What operations need to be done by participants?
All personnel who are authorized to enter the aseptic processing room during manufacturing, including technicians and maintenance personnel, should participate in a media fill at least once a year. Participation should be consistent with the nature of each operator’s duties during routine production.
43. What should be duration of media fill runs?
The duration of aseptic processing operations is a major consideration in media fill design. Although the most accurate simulation model would be the full batch size and duration because it most closely simulates the actual production operations, other appropriate models can be justified. The duration of the media fill run should be determined by the time it takes to incorporate manipulations and interventions, as well as appropriate consideration of the duration of the actual aseptic processing operation. Interventions that commonly occur should be routinely simulated, while those occurring rarely can be simulated periodically.
44. What should be size of media fill runs?
The simulation run sizes should be adequate to mimic commercial production conditions and accurately assess the potential for commercial batch contamination.
The number of units filled during the process simulation should be based on contamination risk for a given process and sufficient to accurately simulate activities that are representative of the manufacturing process. A generally acceptable starting point for run size is in the range of 5,000 to 10,000 units. For operations with production sizes under 5,000, the number of media filled units should at least equal the maximum batch size made on the processing line
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
45. Why high machine speed and low speed should be challenged during media fill runs?
Hgh line speed is often most appropriate in the evaluation of manufacturing processes characterized by frequent interventions or a significant degree of manual manipulation. Use of slow line speed is generally appropriate for evaluating manufacturing processes with prolonged exposure of the sterile drug product and containers/closures in the aseptic area.
46. What types of growth medium should be used during media fill runs?
A microbiological growth medium, such as soybean casein digest medium, should be used.
Use of anaerobic growth media (e.g., fluid thioglycollate medium) should be considered in special circumstances.
47. Which should be characteristic of microbiological growth medium to be used during media fill?
The media selected should be demonstrated to promote growth of gram-positive and gram-negative bacteria, and yeast and mold (e.g., USP indicator organisms).
Environmental monitoring and sterility test isolates can be substituted (as appropriate) or added to the growth promotion challenge.
48. What should be the concentration of organism while testing of Growth promotion units?
Growth promotion units should be inoculated with a <100 CFU challenge.
49. What action should be done if growth promotion test fails?
If the growth promotion testing fails, the origin of any contamination found during the simulation should nonetheless be investigated and the media fill promptly repeated.
50. What should be the incubation duration and temperature for media filled units?
• Incubation temperature should be suitable for recovery of bioburden and environmental isolates and should at no time be outside the range of 20-35 °C. Incubation temperature should be maintained within +2.5 °C of the target temperature.
• Incubation time should not be less than 14 days. If two temperatures are used for the incubation of the media filled units, the units should be incubated for at least 7 days at each temperature (starting with the lower temperature).
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
51. Who should perform inspection of media filled units?
Each media-filled unit should be examined for contamination by personnel with appropriate education, training, and experience in inspecting media fill units for microbiological contamination.
If QC personnel do not perform the inspection, there should be QC unit oversight throughout any such examination.
All suspect units identified during the examination should be brought to the immediate attention of the QC microbiologist.
52. How to perform inspection of media filled units of amber or other opaque containers?
To allow for visual detection of microbial growth, amber or other opaque containers should be substitute with clear containers.
53. After inspection of media filled units which containers shall be incubated and which containers shall not?
All integral units should proceed to incubation. Units found to have defects not related to integrity (e.g., cosmetic defect) should be incubated; units that lack integrity should be rejected.
54. Whether the media filled units generated during the intervention should be incubated or not?
If written procedures and batch documentation are adequate to describe an associated clearance, the intervention units removed during media fills do not need to be incubated. Where procedures lack specificity, there would be insufficient justification for exclusion of units removed during an intervention from incubation. For example, if a production procedure requires removal of 10 units after an intervention at the stoppering station infeed, batch records (i.e., for production and media fills) should clearly document conformance with this procedure. In no case should more units be removed during a media fill intervention than would be cleared during a production run.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
55. How to interpret test results of media fill?
Recommended criteria for assessing state of aseptic line control are as follows:
i. When filling fewer than 5000 units, no contaminated units should be detected.
One (1) contaminated unit is considered cause for revalidation, following an investigation.
ii. When filling from 5,000 to 10,000 units
One (1) contaminated unit should result in an investigation, including consideration of a repeat media fill.
Two (2) contaminated units are considered cause for revalidation, following investigation.
iii. When filling more than 10,000 units
One (1) contaminated unit should result in an investigation.
Two (2) contaminated units are considered cause for revalidation, following investigation.
56. What should be considered when performing investigation of media fill failure?
The microorganisms should be identified to species level.
The investigation should survey the possible causes of contamination. In addition, any failure investigation should assess the impact on commercial drugs produced on the line since the last media fill.
57. To carryout Filtration Efficacy study, which microorganism should be considered for challenge? Why?
The microorganism, Brevundimonas diminuta (ATCC 19146) when properly grown, harvested and used, is a common challenge microorganism for 0.2 μm rated filters because of its small size (0.3 μm mean diameter).
58. What concentration of organism should be considered for Filtration Efficacy study?
A challenge concentration of at least 107 organisms per cm2 of effective filtration area should generally be used, resulting in no passage of the challenge microorganism. The challenge concentration used for validation is intended to provide a margin of safety well beyond what would be expected in production.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
59. What are the factors that can affect filter performance?
Factors that can affect filter performance generally include
(1) Viscosity and surface tension of the material to be filtered,
(2) pH,
(3) Compatibility of the material or formulation components with the filter itself,
(4) Pressures,
(5) Flow rates,
(6) Maximum use time,
(7) Temperature,
(8) Osmolality,
(9) and the effects of hydraulic shock
60. What should be frequency of filter replacement for sterilization filters for product manufacturing?
Sterilizing filters should be routinely discarded after processing of a single lot.
However, in those instances when repeated use can be justified, the sterile filter validation should incorporate the maximum number of lots to be processed.
61. What should be frequency of filter integrity testing?
Integrity testing of the filter(s) can be performed prior to processing, and should be routinely performed post-use. It is important that integrity testing be conducted after filtration to detect any filter leaks or perforations that might have occurred during the filtration.
62. What are the generally used methods for filter integrity testing?
Forward flow and bubble point tests, when appropriately employed, are two integrity tests that can be used.
63. What should be frequency of equipment and accessories sterilization?
Sterility of aseptic processing equipment should normally be maintained by sterilization between each batch.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
64. Why air should be removed from the autoclave chamber while sterilization?
The insulating properties of air interfere with the ability of steam to transfer its energy to the load, achieving lower lethality than associated with saturated steam.
65. Where must to have location for the biological indicator during sterilization validation?
Potentially difficult to reach locations within the sterilizer load or equipment train (for SIP applications) should be evaluated. For example, filter installations in piping can cause a substantial pressure differential across the filter, resulting in a significant temperature drop on the downstream side. We recommend placing biological indicators at appropriate downstream locations of the filter.
66. Why empty chamber mapping studies required during the sterilizer validation?
Empty chamber studies evaluate numerous locations throughout a sterilizing unit (e.g., steam autoclave, dry heat oven) or equipment train (e.g., large tanks, immobile piping) to confirm uniformity of conditions (e.g., temperature, pressure).
67. How heat penetration study shall be done while sterilizer validation?
Heat penetration studies should be performed using the established sterilizer loads. Validation of the sterilization process with a loaded chamber demonstrates the effects of loading on thermal input to the items being sterilized and may identify difficult to heat or penetrate items where there could be insufficient lethality to attain sterility. The placement of biological indicators at numerous positions in the load, including the most difficult to sterilize places, is a direct means of confirming the efficacy of any sterilization procedure. In general, the biological indicator should be placed adjacent to the temperature sensor so as to assess the correlation between microbial lethality and predicted lethality based on thermal input.
68. What types of calibration should be checked which could have impact on sterilization?
i. Temperature and pressure monitoring devices for heat sterilization should be calibrated at suitable intervals. The sensing devices used for validation studies should be calibrated before and after validation runs.
ii. Devices used to monitor dwell time in the sterilizer should be periodically calibrated.
iii. Instruments used to determine the purity of steam (as applicable) should be calibrated. iv. For dry heat depyrogenation tunnels, devices (e.g. sensors and transmitters) used to measure belt speed should be routinely calibrated.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
69. During aseptic area environmental monitoring program, what should be covered?
The environmental monitoring program should cover all production shifts and include air, floors, walls, and equipment surfaces, including the critical surfaces that come in contact with the product, container, and closures.
70. Which location should be covered during environmental monitoring for aseptic area?
It is important that locations posing the most microbiological risk to the product be a key part of the program.
It is especially important to monitor the microbiological quality of the critical area to determine whether or not aseptic conditions are maintained during filling and closing activities. Air and surface samples should be taken at the locations where significant activity or product exposure occurs during production. Critical surfaces that come in contact with the sterile product should remain sterile throughout an operation. When identifying critical sites to be sampled, consideration should be given to the points of contamination risk in a process, including factors such as difficulty of setup, length of processing time, and impact of interventions. Critical surface sampling should be performed at the conclusion of the aseptic processing operation to avoid direct contact with sterile surfaces during processing.
71. How to ensure that environmental monitoring locations are reproducibly monitored?
All environmental monitoring locations should be described in SOPs with sufficient detail to allow for reproducible sampling of a given location surveyed. Written SOPs should also address elements such as (1) frequency of sampling, (2) when the samples are taken (i.e., during or at the conclusion of operations), (3) duration of sampling, (4) sample size (e.g., surface area, air volume), (5) specific sampling equipment and techniques, (6) alert and action levels, and (7) appropriate response to deviations from alert or action levels.
72. Why environmental monitoring results should not be averaged?
Averaging of results can mask unacceptable localized conditions.
73. What should be covered while performing trend analysis of the environmental monitoring data for aseptic area?
Trend reports should include data generated by location, shift, room, operator, or other parameters. Significant changes in microbial flora should be considered in the review of the ongoing environmental monitoring data.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
74. Which are the acceptable methods for monitoring the microbiological quality of the environment in aseptic area?
Acceptable methods for monitoring the microbiological quality of the environment include:
a. Surface Monitoring
b. Active Air Monitoring
c. Passive Air Monitoring (Settling Plates)
75. Which surfaces monitored during Surface Monitoring?
Environmental monitoring involves sampling various surfaces for microbiological quality. For example, product contact surfaces, floors, walls, and equipment should be tested on a regular basis.
76. What types of techniques shall be used for surface monitoring?
Touch plates, swabs, and contact plates can be used for such tests.
77. What types of device shall be used for Active Air Monitoring?
Assessing microbial quality of air should involve the use of active devices including but not limited to impaction, centrifugal, and membrane (or gelatin) samplers.
78. What should be considered during method validation of Passive Air Monitoring (Settling Plates)?
As part of methods validation, the laboratory should evaluate what media exposure conditions optimize recovery of low levels of environmental isolates. Exposure conditions should preclude desiccation (e.g., caused by lengthy sampling periods and/or high airflows), which inhibits recovery of microorganisms.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
79. How microbiological identification is useful in sterile product manufacturing facility environmental monitoring program?
Characterization of recovered microorganisms provides vital information for the environmental monitoring program. Environmental isolates often correlate with the contaminants found in a media fill or product sterility testing failure, and the overall environmental picture provides valuable information for an investigation. Monitoring critical and immediately surrounding clean areas as well as personnel should include routine identification of microorganisms to the species (or, where appropriate, genus) level.
80. How uncontrolled area or lesser controlled areas microbiological identification is useful in sterile product manufacturing facility environmental monitoring program?
In some cases, environmental trending data have revealed migration of microorganisms into the aseptic processing room from either uncontrolled or lesser controlled areas. Establishing an adequate program for differentiating microorganisms in the lesser-controlled environments, such as Class 100,000 (ISO 8), can often be instrumental in detecting such trends.
At minimum, the program should require species (or, where appropriate, genus) identification of microorganisms in these ancillary environments at frequent intervals to establish a valid, current database of contaminants present in the facility during processing (and to demonstrate that cleaning and sanitization procedures continue to be effective).
81. Which method of microbial identification is more accurate?
Genotypic methods have been shown to be more accurate and precise than traditional biochemical and phenotypic techniques. These methods are especially valuable for investigations into failures (e.g., sterility test; media fill contamination). However, appropriate biochemical and phenotypic methods can be used for the routine identification of isolates.
82. What is the goal of microbiological monitoring?
The goal of microbiological monitoring is to reproducibly detect microorganisms for purposes of monitoring the state of environmental control.
83. What should be the capability microbiological culture media used for aseptic area environmental monitoring program?
The microbiological culture media used in environmental monitoring should be validated as capable of detecting fungi (i.e., yeasts and molds) as well as bacteria and incubated at appropriate conditions of time and temperature.
84. What should be incubation condition for environmental monitoring media plates and what should be the duration?
Total aerobic bacterial count can be obtained by incubating at 30 to 35°C for 48 to 72 hours. Total combined yeast and mold count can generally be obtained by incubating at 20 to 25°C for 5 to 7 days.
85. How to ensure that the incoming lots of environmental monitoring media is able to reliably recover microorganisms?
Incoming lots of environmental monitoring media should be tested for their ability to reliably recover microorganisms. Growth promotion testing should be performed on all lots of prepared media. Where appropriate, inactivating agents should be used to prevent inhibition of growth by cleanroom disinfectants or product residuals (e.g., antibiotics).
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
86. Why controlling of Prefiltration Bioburden is important?
Manufacturing process controls should be designed to minimize the bioburden in the unfiltered product. In addition to increasing the challenge to the sterilizing filter, bioburden can contribute impurities (e.g., endotoxin) to, and lead to degradation of, the drug product. A prefiltration bioburden limit should be established.
87. In which scenario alternate Microbiological Test Methods should be used?
Other suitable microbiological test methods (e.g., rapid test methods) can be considered for environmental monitoring, in-process control testing, and finished product release testing after it is demonstrated that the methods are equivalent or better than traditional methods (e.g.,USP).
88. How Particle Monitoring is useful in clean room?
Routine particle monitoring is useful in rapidly detecting significant deviations in air cleanliness from qualified processing norms (e.g., clean area classification).
89. What should be the environmental condition of sterility testing area?
The testing laboratory environment should employ facilities and controls comparable to those used for aseptic filling operations. Poor or deficient sterility test facilities or controls can result in test failure. If production facilities and controls are significantly better than those for sterility testing, the danger exists of mistakenly attributing a positive sterility test result to a faulty laboratory even when the product tested could have, in fact, been nonsterile.
90. Which USP chapter and which part of 21 CFR describe about sterility testing?
Sterility testing methods are required to be accurate and reproducible, in accordance with 211.194 and 211.165.
USP <71> “Sterility Tests” is the principal source used for sterility testing methods, including information on test procedures and media.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
91. What is the probability of detection of contamination during sterility testing?
Sterility tests are limited in their ability to detect contamination because of the small sample size typically used. For example, as described by USP, statistical evaluations indicate that the sterility test sampling plan “only enables the detection of contamination in a lot in which 10% of the units are contaminated about nine times out of ten in making the test” (Ref. 13). To further illustrate, if a 10,000-unit lot with a 0.1 percent contamination level was sterility tested using 20 units, there is a 98 percent chance that the batch would pass the test.
92. How to collect the samples for sterility testing?
It is important that the samples represent the entire batch and processing conditions. Samples should be taken:
• At the beginning, middle, and end of the aseptic processing operation
• In conjunction with processing interventions or excursions
93. When sterility test failure results can be invalidated?
An initial positive test would be invalid only in an instance in which microbial growth can be unequivocally ascribed to laboratory error.
Only if conclusive and documented evidence clearly shows that the contamination occurred as part of testing should a new test be performed.
94. In case of inconclusive investigation, what should be the batch disposition decision?
When available evidence is inconclusive, batches should be rejected as not conforming to sterility requirements.
95. What parameters should be considered while performing sterility failure investigation?
The investigation’s persuasive evidence of the origin of the contamination should be based on at least the following:
1. Identification (speciation) of the organism in the sterility test
2. Record of laboratory tests and deviations
3. Monitoring of production area environment
4. Monitoring Personnel
5. Product Presterilization Bioburden
6. Production record review 7. Manufacturing history
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
96. How isolate identification is useful during investigation of sterility testing failure?
Sterility test isolates should be identified to the species level. Microbiological monitoring data should be reviewed to determine if the organism is also found in laboratory and production environments, personnel, or product bioburden. Advanced identification methods (e.g., nucleic-acid based) are valuable for investigational purposes. When comparing results from environmental monitoring and sterility positives, both identifications should be performed using the same methodology.
97. What is the recommended material of construction for aseptic processing design?
Suitable materials should be chosen based on durability, as well as ease of cleaning and decontamination. For example, rigid wall construction incorporating stainless steel and glass materials is widely used.
98. What is expected classification of isolator and surrounding area?
The interior of the isolator should meet Class 100 (ISO 5) standards. The classification of the environment surrounding the isolator should be based on the design of its interfaces (e.g., transfer ports), as well as the number of transfers into and out of the isolator. A Class 100,000 (ISO 8) background is commonly used based on consideration of isolator design and manufacturing situations. An aseptic processing isolator should not be located in an unclassified room.
Reference: FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice – September 2004
99. What should be the laminar air flow systems air speed for Grade A area? Why?
Laminar air flow systems should provide a homogeneous air speed in a range of 0.36 – 0.54 m/s (guidance value) at the working position in open clean room applications.
100. Why length of tubing and the radii is important for particle monitoring system?
The length of tubing and the radii of any bends in the tubing must be considered in the context of particle losses in the tubing.
101. What should be pressure differential between adjacent rooms of different grades?
Adjacent rooms of different grades should have a pressure differential of 10 – 15 pascals.
102. What is the recommended temperature for Water for injections storage and distribution? Why?
Water for injections should be produced, stored and distributed in a manner which prevents microbial growth, for example by constant circulation at a temperature above 70°C.
103. For what types of areas, fumigation is useful?
Fumigation of clean areas may be useful for reducing microbiological contamination in inaccessible places.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
104. What should be the method of choice for sterilization as per EU Annex-1?
Where possible, heat sterilisation is the method of choice.
105. What should be the verification schedule for sterilization process?
The validity of the sterilization process should be verified at scheduled intervals, at least annually, and whenever significant modifications have been made to the equipment.
106. How to differentiate products which have not been sterilised from those which have?
Each basket, tray or other carrier of products or components should be clearly labelled with the material name, its batch number and an indication of whether or not it has been sterilised. Indicators such as autoclave tape may be used, where appropriate, to indicate whether or not a batch (or sub-batch) has passed through a sterilisation process, but they do not give a reliable indication that the lot is, in fact, sterile.
107. How to ensure temperature during sterilization process?
Each heat sterilisation cycle should be recorded on a time/temperature chart with a sufficiently large scale or by other appropriate equipment with suitable accuracy and precision. The position of the temperature probes used for controlling and/or recording should have been determined during the validation, and where applicable also checked against a second independent temperature probe located at the same position.
108. Can chemical or biological indicator be replacement of physical measurements during sterilization cycle?
Chemical or biological indicators should not take the place of physical measurements.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
109. How to monitor moist heat sterilization cycle?
Both temperature and pressure should be used to monitor the process. Control instrumentation should normally be independent of monitoring instrumentation and recording charts. Where automated control and monitoring systems are used for these applications they should be validated to ensure that critical process requirements are met. System and cycle faults should be registered by the system and observed by the operator. The reading of the independent temperature indicator should be routinely checked against the chart recorder during the sterilisation period. For sterilisers fitted with a drain at the bottom of the chamber, it may also be necessary to record the temperature at this position, throughout the sterilization period. There should be frequent leak tests on the chamber when a vacuum phase is part of the cycle.
110. Explain the quality of wrapping material used during sterilization process?
The items to be sterilised, other than products in sealed containers, should be wrapped in a material which allows removal of air and penetration of steam but which prevents recontamination after sterilisation.
111. What challenge should be carried out during validation when for dry heat sterilization when it is intended to remove pyrogen?
Challenge tests using endotoxins should be used as part of the validation.
112. In what scenario, sterilisation by radiation can be adopted?
Radiation sterilisation is used mainly for the sterilisation of heat sensitive materials and products. Many medicinal products and some packaging materials are radiation-sensitive, so this method is permissible only when the absence of deleterious effects on the product has been confirmed experimentally.
113. Which irradiation is not an acceptable method of sterilisation.?
Ultraviolet irradiation is not normally an acceptable method of sterilisation.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
114. How to measure radiation while doing irradiation serilization?
During the sterilisation procedure the radiation dose should be measured. For this purpose, dosimetry indicators which are independent of dose rate should be used, giving a quantitative measurement of the dose received by the product itself. Dosimeters should be inserted in the load in sufficient number and close enough together to ensure that there is always a dosimeter in the irradiator. Where plastic dosimeters are used they should be used within the time-limit of their calibration. Dosimeter absorbances should be read within a short period after exposure to radiation.
115. How to prevent mix-up between irradiated and nonirradiated materials?
Radiation sensitive colour disks should also be used on each package to differentiate between packages which have been subjected to irradiation and those which have not.
116. When Sterilisation with ethylene oxide should be used?
This method should only be used when no other method is practicable. During process validation it should be shown that there is no damaging effect on the product and that the conditions and time allowed for degassing are such as to reduce any residual gas and reaction products to defined acceptable limits for the type of product or material.
117. What precaution should be taken while sterilization of material using ethylene oxide?
i. Direct contact between gas and microbial cells is essential; precautions should be taken to avoid the presence of organisms likely to be enclosed in material such as crystals or dried protein. The nature and quantity of packaging materials can significantly affect the process.
ii. Before exposure to the gas, materials should be brought into equilibrium with the humidity and temperature required by the process. The time required for this should be balanced against the opposing need to minimize the time before sterilisation.
iii. Each sterilisation cycle should be monitored with suitable biological indicators, using the appropriate number of test pieces distributed throughout the load.
iv. For each sterilisation cycle, records should be made of the time taken to complete the cycle, of the pressure, temperature and humidity within the chamber during the process and of the gas concentration and of the total amount of gas used. The pressure and temperature should be recorded throughout the cycle on a chart.
v. After sterilisation, the load should be stored in a controlled manner under ventilated conditions to allow residual gas and reaction products to reduce to the defined level.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
118. What is preferred method of sterilization as per EU Annex 1?
Filtration alone is not considered sufficient when sterilisation in the final container is possible. With regard to methods currently available, steam sterilisation is to be preferred. If the product cannot be sterilised in the final container, solutions or liquids can be filtered through a sterile filter of nominal pore size of 0.22 micron (or less), or with at least equivalent micro-organism retaining properties, into a previously sterilised container. Such filters can remove most bacteria and moulds, but not all viruses or mycoplasmas. Consideration should be given to complementing the filtration process with some degree of heat treatment.
119. Number of sterilization filters recommended by EU Annex 1 when sterilization is done through filtration method? What should be the location of the filer?
Due to the potential additional risks of the filtration method as compared with other sterilization processes, a second filtration via a further sterilised micro-organism retaining filter, immediately prior to filling, may be advisable. The final sterile filtration should be carried out as close as possible to the filling point.
120. When to perform filter integrity testing and why it is important?
The integrity of the sterilised filter should be verified before use and should be confirmed immediately after use by an appropriate method such as a bubble point, diffusive flow or pressure hold test. The time taken to filter a known volume of bulk solution and the pressure difference to be used across the filter should be determined during validation and any significant differences from this during routine manufacturing should be noted and investigated. Results of these checks should be included in the batch record. The integrity of critical gas and air vent filters should be confirmed after use. The integrity of other filters should be confirmed at appropriate intervals.
121. What is the preferred duration of sterilization filter as per EU Annex 1?
The same filter should not be used for more than one working day unless such use has been validated.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
122. Partially stoppered freeze drying vials should be maintained under which grade area?
Partially stoppered freeze drying vials should be maintained under Grade A conditions at all times until the stopper is fully inserted.
123. At which stage of operation, aseptically filled vial cap is considered integral?
The container closure system for aseptically filled vials is not fully integral until the aluminium cap has been crimped into place on the stoppered vial. Crimping of the cap should therefore be performed as soon as possible after stopper insertion.
124. What is the major risk of vial crimping operation in aseptic area? How to mitigate that risk?
The equipment used to crimp vial caps can generate large quantities of non-viable particulates, the equipment should be located at a separate station equipped with adequate air extraction.
125. Vial capping should be done which grade area?
Vial capping can be undertaken as an aseptic process using sterilised caps or as a clean process outside the aseptic core. Where this latter approach is adopted, vials should be protected by Grade A conditions up to the point of leaving the aseptic processing area, and thereafter stoppered vials should be protected with a Grade A air supply until the cap has been crimped.
126. How many filled containers should be visually inspected? Why?
Filled containers of parenteral products should be inspected individually for extraneous contamination or other defects.
127. Explain the process of visual inspection injectable products?
When inspection is done visually, it should be done under suitable and controlled conditions of illumination and background. Operators doing the inspection should pass regular eye-sight checks, with spectacles if worn, and be allowed frequent breaks from inspection. Where other methods of inspection are used, the process should be validated and the performance of the equipment checked at intervals. Results should be recorded.
Reference: EudraLex – The Rules Governing Medicinal Products in the European Union Volume 4, EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex 1 – Manufacture of Sterile Medicinal Products – 01 March 2009
Top 10 important Interview Questions and Answers on Pharmaceutical Validation

This page covers most of the interview questions and answers asked during a technical interview round of quality assurance and validation professionals.
You will find interview questions and answers on Terminologies associated with process validation, Stages of Process Validation, approach to process validation, Stages of process validation, typical steps for QbD, control strategy of process validation, FDA guidance, EMA guidance, WHO guidance on hold time studies of the products, different guidelines/ regulations describing requirement of cleaning validation, and different guidelines/ regulations describing requirement of equipment qualification.
The interview questions cover questions from basic to advance level of technical aspects. These interview questions and answers will help to crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
The topics covered here are the sterilization process, aseptic processing, media fill, area classification, and associated topics. In addition, the interview questions and answers cover various equipment used for the manufacturing process of sterile formulations.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning, and best of luck with your interview! Happy Learning.
1. Terminologies associated with process validation.
i. Attribute
An attribute is a physical, chemical, or microbiological property or characteristic of an input or output material.
Reference: ICH Quality Guideline Q5E
ii. Critical Quality Attribute (CQA)
A CQA is a physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
Reference: ICH Quality Guideline Q8 (R2)
iii. Quality Attribute
A Quality Attribute is a molecular or product characteristic that is selected for its ability to indicate the quality of the product. Collectively, the quality attributes define identity, purity, potency and stability of the product, and safety with respect to adventitious agents. Specifications measure a selected subset of the quality attributes.
Reference: ICH Quality Guideline Q6B
iv. Control Strategy
A control strategy is a planned set of controls, derived from current product and process understanding that assures process performance and product quality (ICH Q10).
Every drug substance manufacturing process, whether developed through a traditional or an enhanced approach (or some combination thereof), has an associated control strategy.
A control strategy can include, but is not limited to, the following:
Controls on material attributes (including raw materials, starting materials, intermediates, reagents, primary packaging materials for the drug substance, etc.);
Controls implicit in the design of the manufacturing process (e.g., sequence of purification steps (Biotechnological/Biological Products), or order of addition of reagents (Chemical Products));
In-process controls (including in-process tests and process parameters); Controls on drug substance (e.g., release testing).
Reference: ICH guideline Q11
v. Continued Process Verification (CPV)
The CPV is the Stage 3 of Process Validation. The goal of this stage is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture.
This is science and risk-based approach for collection and evaluation of information and data about the performance of the process, which will allow detecting undesired process variability. Evaluating the performance of the process identifies problems and determines whether action must be taken to correct, anticipate, and prevent problems so that the process remains in control (§ 211.180(e)).
vi. Critical process parameter (CPP):
A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality.
Reference: ICH guideline Q8
vii. Critical Material Attribute (CMA)
Critical material attribute (CMA) are defined as A material whose variability (physical, chemical, biological or microbiological property or characteristic of an input material) has an impact a critical quality attribute and therefore it should be monitored or controlled to ensure desired drug product quality.
Reference: http://www.rsc.org/
viii. Design Space
The design space is the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality.
Working within the design space is not considered a change. Movement out of the design space is considered to be a change, and would normally initiate a regulatory post-approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.
Reference: ICH guideline Q8 (R2)
ix. Formal Experimental Design (synonym: design of experiments)
A Formal Experimental Design is a structured, organized method for determining the relationship between factors affecting a process and the output of that process.
Reference: ICH guideline Q8 (R2)
x. Lifecycle
Lifecycle includes all phases in the life of a product, from the initial development through marketing until the product’s discontinuation.
Reference: ICH guideline Q8 (R2)
xi. Normal Operating Range (NOR)
The NOR is a defined range, within (or equal to) the Proven Acceptable Range, specified in the manufacturing instructions as the target and range at which a process parameter is controlled, while producing unit operation material or final product meeting release criteria and CQAs.
Reference: Process Robustness – A PQRI White Paper, Pharma. Engin. 2006.
xii. Key Process Parameter (KPP; synonym: key operational parameter)
This is an input process parameter that should be carefully controlled within a narrow range and is essential for process performance. A key process parameter does not affect product quality attributes. If the acceptable range is exceeded, it may affect the process (e.g., yield, duration) but not product quality.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xiii. Non-Key Process Parameter (Non-KPP; synonym: non-key operational parameter)
This is an input parameter that has been demonstrated to be easily controlled or has a wide acceptable limit. Non-key operational parameters may have an impact on quality or process performance if acceptable limits are exceeded.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xiv. Process Parameter (synonym: operational parameter)
This is an input variable or condition of the manufacturing process that can be directly controlled in the process. Typically, these parameters are physical or chemical (e.g., temperature, process time, column flow rate, column wash volume, reagent concentration, or buffer pH).
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xv.Platform Manufacturing
This means the development of a production strategy for a new drug starting from manufacturing processes similar to those used to manufacture other drugs of the same type (the production for which there already exists considerable experience).
Reference: ICH guideline Q11
xvi.Process Analytical Technology (PAT)
A PAT is a system for designing, analyzing, and controlling manufacturing through timely measurements (i.e., during processing) of critical quality and performance attributes of raw and in-process materials and processes with the goal of ensuring final product quality.
Reference: ICH guideline Q8 (R2)
xvii.Process Performance Qualification (PPQ)
This is the second element of Process Qualification. It includes a combination of the actual facility, utilities, equipment, and trained personnel with the commercial manufacturing process, control procedures, and components to produce commercial batches. A successful PPQ will confirm the process design and demonstrate that the commercial manufacturing process performs as expected. Batches prepared are also called ‘Conformance batches’ or ‘PPQ batches’.
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
xviii.Process Qualification
This qualification confirms that the manufacturing process, as designed, is capable of reproducible commercial manufacturing..
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
It consists of following 2 important elements:
(i) Qualification of Facility, Equipment, and Utilities
(ii) Process Performance Qualification
xix. Process Robustness
Ability of a process to tolerate variability of materials and changes of the process and equipment without negative impact on quality is known as process robustness.
Reference: ICH guideline Q8 (R2)
xx. Process Validation
As per US FDA
Such validation is the collection and evaluation of data from the process design stage to commercial production, which establishes with scientific evidence that a process is capable of consistently delivering quality products.
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
As per EMA
Such validation comprises documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes.
Reference: Draft Guideline on Process Validation, MA/CHMP/CVMP/QWP/70278/2012-Rev1; European Medicines Agency: 2012.
xxi. Prospective approach to PPQ
This indicates an approach wherein the Process Performance Qualification batches, manufactured using a qualification protocol, are released for distribution only after complete execution of the Process Performance Qualification Study
Reference: ISPE Guidance
xxii. PPQ re-verification
This indicates the repeating of a part of or a complete PPQ study in the event of changes in the process, equipment, etc. or as a recommendation of the CPV process to verify whether a process continues in a validated state of control and/or to verify that the changes do not adversely impact process characteristics and product quality or the validated state of control of the process
Reference: ISPE Guidance
xxiii. Product Lifecycle
All phases of product stats from the initial development through marketing until the product discontinuation.
xxiv. Process Validation Master Plan (synonym: validation master plan)
This is a document that defines the process validation scope and rationale and that contains the list of process validation studies to be performed.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xxv. Quality
This indicates the suitability of either a drug substance or drug product for its intended use. This term includes such attributes as the identity, strength and purity.
Reference: ICH guideline Q6A
xxvi. Quality by Design (QbD)
This means a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
Reference: ICH guideline Q8 (R2)
xxvii. Quality Target Product Profile (QTPP)
QTPP is a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product.
Reference: ICH guideline Q8 (R2)
xxviii. Verification
Verification is a systematic approach to verify that manufacturing systems, acting alone or in combination, are fit for intended use, have been properly installed, and are operating correctly. This is an umbrella term that encompasses types of approaches to ensure that the systems are fit for the designed purpose. Other terms used are qualification, commissioning and qualification, system validation, etc.
Reference: ASTM E2500-07. Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment; American Society for Testing and Materials: 2007.
xxix. Worst Case
A set of conditions encompassing upper and lower processing limits and circumstances, including those within standard operating procedures, that pose the greatest chance of process or product failure (when compared to ideal conditions). Such conditions do not necessarily induce product or process failure.
Reference: EudraLex: The Rules Governing Medicinal Products in the European Union: Volume 4 Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex. 15.
Qualification and Validation; European Commission: 2014.
2. What are the Stages of Process Validation?
Stage 1 – Process Design
Stage 2 – Process Qualification
Stage 3 – Continued Process Verification
3. What is an integrated team approach to process validation?
Integrated team approach to process validation that includes expertise from a variety of disciplines (e.g., process engineering, industrial pharmacy, analytical chemistry, microbiology, statistics, manufacturing, and quality assurance).
4. Explain “Process Design”, Stage 1 of process validation.
Objective: To design a process that can consistently deliver a commercial product meeting quality attributes.
a. Building and Capturing Process Knowledge and Understanding from
• Previous experience with similar processes
• Product and process understanding (from clinical and pre-clinical activities)
• Analytical characterization
• Published literature
• Engineering studies/ batches
• Clinical manufacturing
• Process development and characterization studies
• Product development activities
• Design of Experiment (DOE) studies to develop process knowledge and relationships between the variable inputs (e.g., component characteristics 13 or process parameters) and the resulting outputs (e.g., in-process material, intermediates, or the final product)
• Computer-based or virtual simulations of certain unit operations or dynamics can provide process understanding and help avoid problems at commercial scale
• Documentation of process understanding, activities and studies
b. Establishing a Strategy for Process Control
An appropriate control strategy is based on knowledge and experience gained in Stage 1 that will help to control the manufacturing process.
Strategy for Process Control includes the following elements:
• Raw material controls
• In-process and release specifications
• In-process controls
• Performance parameters
• Process parameter set points and ranges
• Process monitoring (data review, sampling, testing)
• Processing and hold times
• Process Analytical Technology (PAT)
5. In which scenarios process control through operational limits and in-process monitoring is essential?
In case of following two possible scenarios, process to be controlled using operational limits and in-process monitoring:
a. When the product attribute is not readily measurable due to limitations of sampling or detectability (e.g., viral clearance or microbial contamination) or
b. When intermediates and products cannot be highly characterized and well-defined quality attributes cannot be identified.
6. Explain “Process Qualification”, Stage 2 of process validation.
Objective: In this stage, the process design is evaluated to determine if it is capable of reproducible commercial manufacture.
This stage has two elements:
i. Design of the facility and qualification of the equipment and utilities
ii. Process performance qualification (PPQ).
a. PPQ Protocol
b. PPQ Protocol Execution and Report
CGMP-compliant procedures must be followed. Successful completion of Stage 2 is necessary before commercial distribution. Products manufactured during this stage, if acceptable, can be released for distribution.
7. Explain “Continued Process Verification”, Stage 3 of process validation.
Objective: The goal of the third validation stage is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. A system or systems for detecting unplanned departures from the process as designed is essential to accomplish this goal.
8. Explain typical steps for QbD steps.
9. What is Quality Target Product Profile (QTPP)? Explain with examples.
The QTPP is defined by capturing all relevant quality requirements of the drug product to be developed.
It consists of following:
i. Dosage form and strength, route of administration, delivery systems, container and closure system etc.
ii. Drug substance quality attributes required for intended drug product, i.e. physical, chemical, and biological properties
iii. Drug product quality attributes required for dosage form, i.e. physical, chemical and microbiological attributes of drug products.
iv. Bioavailability attributes, i.e. dissolution requirement or other relevant characteristics. v. Excipient quality attributes, input material compatibility, stability, pharmacology, etc.
10. What is a control strategy? What shall be included while defining control strategy?
• Raw material controls
• In-process and release specifications
• In-process controls
• Performance parameters
• Process parameter set points and ranges
• Process monitoring (data review, sampling, testing)
• Processing and hold times
• Process Analytical Technology (PAT)
11. Which FDA guidance or Code of Federal Regulation suggests having hold time studies of the products?
Establishing production time limits is an example of a control to prevent growth of objectionable microorganisms. Per 21 CFR 211.111, time limits for the completion of each phase of production, when appropriate, must be established and followed. For example, if a firm finds it necessary to hold a bulk topical or liquid product for several months until it is filled, the firm might establish a holding time limit to help prevent objectionable microbial buildup. Validation and control over microbial content of purified water systems used in certain topical products are also examples of such procedures (see FDA guidance, referenced below).
References:
21 CFR 211.113: Control of microbiological contamination
21 CFR 211.165: Testing and release for distribution
21 CFR 211.111: Time limitations on production
FDA Guidance for Industry, 2011, Process Validation: General Principles and Practices
12. Which EMA guidance suggests having hold time studies of the products?
4 July 2017, EMA/CHMP/QWP/245074/2015, Committee for Human Medicinal Products (CHMP), Guideline on manufacture of the finished dosage form
As per guideline
“Depending on the nature of the process and the product (e.g. sterile products), manufacturing durations of critical steps and hold times should be stated and justified”.
“Where relevant, the maximum holding times of the bulk product or, alternatively, the maximum batch manufacturing time from start of product manufacture to completion of packaging into the final primary container for marketing should be stated, appropriately justified and supported by data in relevant parts of the dossier (e.g. challenging the maximum hold time in process validation studies or providing dedicated stability studies for the bulk storage)”.
13. Which WHO guidance suggests having hold time studies of the products?
WHO Technical Report Series No. 992, 2015, Annex 4, General guidance on hold-time studies.
As per guidance
“Normally, intermediate and bulk products should not be stored beyond the established hold time. The choice of maximum holding period should be supported by relevant data. Studies may extend beyond the chosen maximum but it is not necessary to extend testing to determine the extreme limits at which failure occurs”.
14. What are the different guidelines/ regulations describing requirement of cleaning validation?
A. U.S. Food and Drug Administration (U.S. FDA)
PART 211 — CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS – Subpart D – Equipment
Source: Sec. 211.67 Equipment cleaning and maintenance.
Questions and Answers on Current Good Manufacturing Practices – Equipment
GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES
Source: https://www.fda.gov/validation-cleaning-processes-793
B. European Medicines Agency (EMA)
20 November 2014, EMA/CHMP/ CVMP/ SWP/169430/2012, Committee for Medicinal Products for Human Use (CHMP), Committee for Medicinal Products for Veterinary Use (CVMP), Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities
19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018 Committee for Medicinal Products for Veterinary Use (CVMP) Committee for Medicinal Products for Human Use (CHMP) Questions and answers on implementation of risk-based prevention of cross-contamination in production and ‘Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities’ (EMA/CHMP/CVMP/SWP/169430/2012)
C. World Health Organization (WHO)
QAS20 849 Points to consider on the different approaches –including HBEL – to establish carryover limits in cleaning validation for identification of contamination risks when manufacturing in shared facilities
WHO good manufacturing practices for active pharmaceutical ingredients
Source: https://www.who.int/medicines/areas/quality_safety/quality_assurance/GMPActivePharmaceuticalIngredientsTRS957Annex2.pdf
Quality assurance of pharmaceuticals: A compendium of guidelines and related materials
Source: https://www.who.int/medicines/areas/quality_safety/quality_assurance/QualityAssurancePharmVol2.pdf
Points to consider when including6 Health-Based Exposure Limits (HBELs) in cleaning validation
D. Pharmaceutical Inspection Co-operation Scheme (PIC/S)
Cross-contamination in shared facilities (PI-043-1)
Source: https://picscheme.org/docview/2270
Guideline on Setting HBEL for use in risk identification in the manufacture of different medicinal products in shared facilities (PI 046-1)
Source: https://picscheme.org/docview/2467
Guide to GMP for medicinal products Part 1 (PE 009-14 (Part I))
Source: https://picscheme.org/docview/4205
Inspection of Health Based Exposure Limit (HBEL) Assessment and use in Quality Risk Management (PI 052-1)
Source: https://picscheme.org/docview/1947
E. Therapeutic Goods Administration (TGA)
The TGA is adopting version PE009-13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products (PIC/S Guide to GMP)
Source: https://www.tga.gov.au/sites/default/files/transition-new-gmp-requirements-medicinal-products.pdf
TGA interpretation and expectations for demonstrating compliance
Source: https://www.tga.gov.au/resource/pe009-pics-guide-gmp-medicinal-products
A presentation on Cleaning Validation by TGA
Source: https://www.tga.gov.au/sites/default/files/presentation-cleaning-validation.pdf
F. Health Canada
Cleaning validation guide (GUI-0028)
G. Active Pharmaceutical Ingredients Committee (APIC)
Guidance on aspects of Cleaning Validation in Active Pharmaceutical Ingredient Plants APIC Cleaning Validation 2016
Source: https://apic.cefic.org/pub/APICCleaningValidationGuide-updateSeptember2016-final.pdf
Guidance on aspects of Cleaning Validation in Active Pharmaceutical Ingredient Plants APIC Cleaning Validation 2021
Source: https://apic.cefic.org/publications/APIC_Cleaning-validation-guide_2021.pdf
H. Parenteral Drug Association (PDA)
PDA Technical Report 29: Points to Consider for Cleaning Validation
PDA Technical Report 49: Points to Consider for Biotechnology Cleaning Validation
I. International Society For Pharmaceutical Engineering (ISPE)
Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products 2nd Edition ISPE Risk-MaPP
Cleaning Validation Lifecycle – Applications, Methods, and Controls ISPE Cleaning Validation Guideline
J. American Society For Testing and Materials (ASTM)
ASTM E3106 – 18e1 (Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation)
Standard Guide for Derivation of Health-Based Exposure Limits (HBELs) ASTM E3219
15. What are the different guidelines/ regulations describing requirement of equipment qualification?
a. EU GMP Annex 15 “Qualification and Validation”, Eudralex 2015
b. PIC/S GMP for Medicinal Products Annex 15 “Qualification and Validation” PE009-14 (Annexes) 2018
c. PE 009-15 (Annexes) ANNEX 15 – Qualification and validation
d. PI 006-3 – Validation Master Plan installation and operational qualification non-sterile process validation cleaning validation
e. Subpart C – Building and Facilities Section 211.42 – Design and Construction Features
f. USP 37: “Analytical Instrument Qualification”, (section 1058), The United States Pharmacopeial Convention, 2016
g. ISPE Baseline Guide Volume 5: “Commissioning & Qualification”, Second Edition, 2019
h. ASTM E2500: “Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment”, ASTM 2020
i. ISPE Good Practice Guide: Applied Risk Management for Commissioning and Qualification, ISPE 2011
j. PDA Technical Report 54-5: “Quality Risk Management for the Design, Qualification, and Operation of Manufacturing Systems”, PDA 2017
k. EU GMP Annex 11 “Computerised Systems”, Eudralex 2011
l. US FDA Guidance for Industry: “Process Validation: General Principles and Practices, 2011
m. ISPE GAMP® 5 Guide: “A Risk-Based Approach to Compliant GxP Computerized Systems”, ISPE 2008
n. ISPE Guide: “Science and Risk-Based Approach for the Delivery of Facilities, Systems, and Equipment”, ISPE 2011
o. FDA US CFR 21 part 11 Electronic Records; Electronic Signatures 2003
p. Health Canada: Guide to validation – drugs and supporting activities (GUI-0029)
q. WHO Technical Report Series, No. 937, Annex 4: Supplementary guidelines on good manufacturing practices: validation
Top 70+ Interview Questions for Pharmaceutical Microbiology

This page covers most of the interview questions and answers asked during a technical interview round of Pharmaceutical Microbiology.
You will find interview questions and answers on microbiology basics, pharmacopoeial chapter and sections applicable for microbiology, Culture Media, Growth Promotion test, microbiological quality control for sterile and non-sterile dosage forms, good practice in a microbiological laboratory, aseptic technique to be followed in the microbiological laboratory, pure culture, Pyrogens and endotoxins, different staining techniques used in the pharmaceutical microbiology laboratory, microbial identification, Gram-staining technique, fungal staining, approved culture collections, sterility testing and environmental monitoring.
The interview questions cover questions from basic to advance level of technical aspects. These interview questions and answers will help to crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning, and best of luck with your interview! Happy Learning.
1. Explain the parts of compound light microscopes used in the pharmaceutical microbiology laboratory?

● Illuminator: Light source at the base of the microscope enable better view of object;
● Condensor: This is a two lens system that collects and concentrates light from the illuminator and directs it to the iris diaphragm;
● Iris diaphragm: regulates the amount of light entering the lens system;
● Stage: This is a platform for the slide with hole in the center to let light from the illuminator pass through. It consists of clips to hold the slide lace;
● Eye pieces: To view object;
● Objectives: To magnify the object as per rating on it
2. What are the pharmacopoeial chapter and sections applicable for microbiology?
| Pharmacopoeial chapter(s) | Use |
| Microbiological examination of nonsterile products: total viable aerobic count (Ph. Eur. 2.6.12, USP <61>) USP <2021> Microbial enumeration tests-nutritional and dietary supplements USP <2023> Microbiological attributes of nonsterile nutritional and dietary supplements | Organism count in raw materials, water, finished products |
| Microbiological examination of nonsterile products: tests for specified organisms (Ph. Eur. 2.6.13, USP <62>) USP <2022> Microbiological procedures for absence of specified microorganismsnutritional and dietary supplements | Type of organisms present in raw materials, water, finished products |
| Ph. Eur. 5.1.4 Microbiological quality of pharmaceutical preparations/USP <1111> Microbiological attributes of nonsterile pharmaceutical products | Determining limits and control factors |
| Sterility (Ph. Eur. 2.6.1, USP <71>) Pyrogens/endotoxin Rabbit Pyrogen Test (Ph. Eur. 2.6.8, USP<151>) Limulus amoebocyte lysate (LAL) bacterial endotoxin test (Ph. Eur. 2.6.14, USP<85>) | Sterility test for finished products endotoxin test for raw materials, pharmaceuticals waters, finished products |
| Antimicrobial preservative efficacy testing (Ph. Eur. 5.1.3, USP <51>) | Antimicrobial preservative efficacy |
| Microbiological assay of antibiotics (E.P 2.7.2., USP<81>) | Assays of antibiotic |
| USP <1112> Application of water activity determination to nonsterile pharmaceutical products | Water activity can be used to predict microbial proliferation in the product |
| USP <1211> Sterilization and sterility assurance of compendial articles. There are a series of subchapters that describe specific sterilization methods | Sterilization and microbial reduction |
| USP <55> Biological indicators | Biological indicators for assessing microbial reduction |
| USP <1113> Microbial characterization, identification, and strain typing | Microbial identification |
| USP <1117> Microbiological best laboratory practice | Management of microbiological good laboratory practice |
| USP <1116> Microbiological control and monitoring of aseptic processing environments | Environmental monitoring for aseptic environments |
| Ph. Eur. 5.1.6 Alternative methods for control of microbiological quality/ USP <1223> Validation of alternative microbiological methods | Alternative methods/ Rapid microbiological methods |
| USP <1115> Bioburden control of nonsterile drug products | Bioburden control |
| USP <1227> Validation of microbial recovery from pharmacopeial articles | Microbial method validation |
3. Which factors of culture media affects the cultivation of microorganisms?
Culture media factors affecting the cultivation of microorganisms are optimum nutrients, oxygen or other gases, moisture, pH, and temperature.
4. What are the important nutrients of culture media that affects the cultivation of microorganisms?
The key nutrients of culture media that affects the cultivation of microorganisms are sources of carbon, nitrogen, water, inorganic phosphates and sulfur, vitamins, and trace metals.
5. Whose name recognize as “The Father of Culture Media”?
Robert Koch (1843–1910) discovered that broths based on fresh beef serum or meat extracts (so-called bouillons, the term “broth” for liquid culture medium being analogous to broth or soup) produced optimal growth.
6. Who was the first scientist cultivated the microorganisms on a growth medium?
The French chemist and microbiologist, Louis Pasteur (1922–1985).
7. Petri dish is named based on which scientists’ contribution?
Julius Richard Petri (1852–1921)
8. What are the different types of culture medium used in pharmaceuticals and what is the purpose of those?
| Types of medium | Use |
| Nutrient agar or broth and tryptone soya agar or broth. Tryptone soya agar (equivalent to soyabean casein digest medium) | → Environmental monitoring→ Isolation and cultivation of nonfastidious and fastidious microorganismsNote: Fastidious bacteria are bacteria that need special nutritional supplements and conditions to grow on agar plates. Nonfastidious bacteria are bacteria that do not need special nutritional supplements and conditions to grow on agar plates |
| Tryptone soya broth | Sterility testing and as a general growth broth in microbial enumeration tests, as well as used for media simulation trials |
| vegetable peptone broth | Media filling trials |
| fluid thioglycollate medium | used for the growth of bacteria (aerobic and anaerobic) as a part of the sterility test |
| fungi | Sabouraud dextrose agar or malt extra agar |
| R2A | Microbiological examination of water (This is a low nutrient agar used for the cultivation of heterotrophic microorganisms). |
| Columbia blood agar | Detection of hemolytic reactions by Staphylococci |
9. What is the process of Growth Promotion test of microbiology culture media? What should be the acceptance criteria?
1. Medium to be inoculated with a microorganisms <100 Colony Forming Units (CFU).
2. Use not less than five unique strains as recommended by the pharmacopoeias and two organisms from environment isolates on rotation basis (It should be within the five passages from the original reference culture seed lot).
3. Compare the growth on the medium with medium previously approved culture lot.
4. Acceptance is no more than a factor of 2 differences in productivity ratio calculations.
10. What is the calculation of productivity ratio and what is the acceptance criterion for agar media plate or solid media for growth promotion test?
Productivity ratio =
Mean of two test plates (cfu)/ Mean of two comparative control plates (cfu)
An acceptable productivity ratio should be (0.5 – 2.0). That is equivalent to a 50 – 200 % recovery.
11. What are the quantitative techniques of Growth Promotion Tests for Solid media?
Quantitative techniques of Growth Promotion Tests are of two types.
● Ecometric
● Miles – Misra (Drop Count)
12. Explain ecometric method of Growth Promotion Tests?
This technique is semi-quantitative variant of the streaking.
This technique is operator-dependent and has lower precision. However, it can be used to great effect with practice.
Following are the steps:
● A fresh suspension of the challenge organism is taken into a calibrated loop (One loopful of inoculm).
● Five streaks are streaked out into four quadrants onto the agar plate along with a final streak in the center of the plate.
● These plates are then incubated overnight for growth.
● The patterns of growth are interpreted to provide an Absolute Growth Index (AGI)
13. Explain Miles – Misra technique (the drop count technique) of Growth Promotion Tests?
This technique involves spreading droplets of known quantities (10 μL) of microbial suspensions.
The test plate is compared against control plate after incubation to verify number of colonies recovered.
Note:
The accuracy of the method is dependent on following factors:
● Dilution used
● Number of colony forming units (cfu) in the inoculum
● Volume of the inoculums
● Spreading technique
14. What are the quantitative techniques of Growth Promotion Tests for Broth Media?
Quantitative techniques of Growth Promotion Tests for Broth Media are of four types.
● Copious Growth
● End-point Methods
● Most Probable Number (MPN)
● Kinetic Parameters
15. Explain Copious Growth method of Growth Promotion Tests for Broth Media?
This is the method of choice as per compendial. The challenge of broth media is done by comparing the growth over the period of time with a control batch (which provides a qualitative assessment of copious growth). In this method, the challenge organism is inoculated at a very low level (< 100 CFU per unit) and incubated at the prescribed temperature for the prescribed period of time (3 days or 5 days).
The advantage of this method is that it does not require a great deal of labor. Semi-quantitative assessment can be done by constructing a growth index from slight to copious growth (normally a scale of +, ++, or +++).
16. Explain End-point Methods method of Growth Promotion Tests for Broth Media?
In this method, very low levels of inoculum are added to multiple tubes of the two media under testing. Growth frequency is compared between the two media to understand the equivalency.
17. Explain Most Probable Number (MPN) method of Growth Promotion Tests for Broth Media?
This is a Microbial Limits Test. In this method, the unknown sample is prepared in a ten-fold dilution series and added to nutrient broth in replicate tubes. The tubes will then either turn turbid (growth) or remain clear, and allow for an estimate of the most probable number of microorganisms.
18. Explain Kinetic Parameters method of Growth Promotion Tests for Broth Media?
The growth promoting testing of two lots of broth can be compared by measuring the growth curves of same inocula grown side-by-side. The growth rate of an organism can be determined by spectrophotometrically or by viable count.
19. How Growth Promotion Tests for selective media is performed?
Selective media requires a different approach than general purpose media. The objective of the test for selective media is to see that the media supports the growth of specific microorganisms. Inhibition, colony morphology and pigment are verified during the testing. In order to determine growth of selective media, specific microorganisms are used as positive or negative indicators.
20. What are the steps involved in the manufacturing of culture media?
a. Collection of prerequisite
● Decide batch size
● Vessel for holding or dispensing the media
● Weighing balance
● Petri dishes/ Containers
● Dehydrated media
b. Rehydration of media
● Media powder is rehydrated by mixing the medium in the volume of water (Amount should be based on manufacturer’s instruction)
● Homogenize the solution by mixing
● Care must be taken to avoid scorching the media.
● The media should clarify near boiling (95–100 °C), and the media should only be allowed to boil less than 1 min.
c. Sterilization
d. Addition of supplements (depending on the types of media requirement)
e. Filling in container
f. Status labeling
21. What are the acceptance criteria for microbiological quality of non-sterile dosage forms and raw materials?
| Application | TAMC | TYMC | Specified microorganisms |
| Non-aqueous preparations for oral use | 1000 | 100 | Escherichia coli absent in 1 g or 1 mL |
| Aqueous preparations for oral use | 100 | 10 | Escherichia coli absent in 1 g or 1 mL |
| Rectal use | 1000 | 100 | Need based |
| Oromucosal use Gingival use Cutaneous use Nasal use Auricular use | 100 | 10 | Staphylococcus aureus absent in 1 g or 1 mL Pseudomonas aeruginosa absent in 1 g or 1 mL |
| Vaginal use | 100 | 10 | Staphylococcus aureus absent in 1 g or 1 mL Pseudomonas aeruginosa absent in 1 g or 1 mL Candida albicans absent in 1 g or 1 mL |
| Inhalation use | 100 | 10 | Staphylococcus aureus absent in 1 g or 1 mL Pseudomonas aeruginosa absent in 1 g or 1 mL Bile-tolerant gram-negative bacteria absent in 1 g or 1 mL |
| Transdermal patches | 100 | 10 | Staphylococcus aureus absent in 1 g or 1 mL Pseudomonas aeruginosa absent in 1 g or 1 mL |
| Raw material for pharmaceutical use | 1000 | 100 | Need based |
22. What are the risks because of microorganism in the pharmaceutical formulation if not within the acceptable limit?
● Decomposition of the product
● Degradation of product
● Infection to the patient
23. Give few examples of good practice in a microbiological laboratory.
● Required equipment and media should sterilized prior to use.
● Sterilized equipment and media should be maintained and stored in such a way that it should not be contaminated.
● Frequently disinfect hands and working surfaces while carryout aseptic operations.
● Fly, rodent and pest control.
● Use personnel protective equipment such as laboratory coat, safety glasses, and gloves.
● Drinkable and eatables should not be allowed in the laboratory.
● Sterilize contaminated used media and waste prior to disposal.
● Pipetting should not be done mouth.
24. Why aseptic technique should be followed in the microbiological laboratory?
To prevent contamination and cross-contamination in microbiology laboratory. Probability of contamination in the laboratory is – samples, media, environment, facility etc.
25. How asepsis can be achieved in the laboratory?
● Frequent washing and disinfecting hands.
● Use of unidirectional airflow station for critical operations.
● Handling of positive control at the end of analysis.
● Not touching the samples and accessories directly by hand.
26. What is pure culture?
A pure culture is a population of cells or multicellular organisms growing in the absence of other species or types in which cells are genetic clones of one another.
27. What is most common method to isolate individual cells and produce a pure culture?
The most common method to isolate individual cells and produce a pure culture is to prepare a streak plate method.
28. Explain Streak Plate Method.
a. Sterilize the inoculating loop by heating it until red hot in a flame until it is red hot. Allow it to cool.
b. Pick up a loop full of liquid inoculum or bacterial growth from the surface of an agar plate and, starting about 2.5 cm in from the edge of the plate, streak lightly back and forth with the loop flat, making close, parallel streaks back to the edge of the plate to a quarter of the plate.

c. Sterilize the loop and cool again, going back to the edge of area first quarter that you just streaked, extend the streaks into the second quarter of the plate.
d. Repeat the same process for remaining two quarter of the plate.
e. Flame and cool the loop again.
29. What should be done during microbiology testing if sample exhibit antimicrobial activity?
When the sample possesses antimicrobial activity, it that requires neutralization. Following are commonly used principle for neutralization:
● Chemical neutralization
● Enzymatic neutralization
● Dilution
30. What is Pyrogens and endotoxins?
Pyrogens and endotoxins are a heterogeneous group of chemical entities that cause fever when injected.
31. What is the source of pyrogens and endotoxins?
Pyrogens can be nonbacterial as well as bacterial in origin.
In the pharmaceutical industry, mostly observed source is from Gram-negative bacteria. That is the lipopolysaccharide (LPS) from the bacterial cell wall.
Pyrogens are the metabolic byproduct of the Gram-negative bacteria.
32. Explain types of microbial identification methods.
Identification methods can be divided into two types:
● Phenotypic
● Genotypic
33. What are the differences between Phenotype and Genotype?
● “Genotype” is an organism’s full hereditary information.
● “Phenotype” is an organism’s actual observed properties, such as morphology, development or behavior.
34. What are the different staining techniques used in the pharmaceutical microbiology laboratory?
● Gram-stain
● Bacterial spore stain
● Fungal staining
● Ziehl–Neelsen stain
35. What is the objective of microbial identification?
The objective of microbial identification is to differentiate one microbial isolate from another with respect to family (genus) and a species or a particular strain.
36. What are the taxonomic terms for microbial identification?
● Family: group of similar genera
● Genus: group of similar species
● Species: group of similar strains
● Type: strains within a species
● Strain: an isolate of a particular species
37. What is the first step of microbial identification?
The first step of most identification program is colony and cellular morphology of the microorganism.
Colony morphology is a method that used to describe the characteristics of an individual colony of bacteria on agar media in a Petri dish.
Colony morphology is normally classified based on the form, elevation and margin. This can be further classified as follows.

38. What is Gram-stain?
The Gram stain is an important technique for identification of bacteria. It divides bacteria into two groups, Gram-positives and Gram-negatives.
39. How the Gram-staining technique is useful?
Gram-staining technique that allows to visualize the morphological types of bacteria using a compound light microscope under magnification of 100x.
40. Explain the principle of the Gram-staining technique?
Step 1: Crystal violet (primary stain) aqueous solutions consist of +ve and –ve ions. These ions penetrate through the cell wall and cell membrane of all types of bacteria. The +ve ion stains the bacterial cells and stains the cells purple.
Step 2: Iodide (the mordant) interacts with crystal violate and forms complexes.
Step 3: Decolorizer (made of acetone and alcohol 95%) interacts with the lipids of the cell membrane. A Gram-negative cell loses its outer lipopolysaccharide membrane, and the inner peptidoglycan layer is left exposed.
On the other hand, a Gram-positive cell becomes dehydrated because of property of decolorizer. The complexes trapped within the Gram-positive cell due to the multilayered nature of the peptidoglycan.
Step 4: When decolorization is added, the Gram-positive cell remains purple, and the Gram-negative cell loses its purple color.
Step 5: A Safranin, the counterstain is used to provide color to Gram-negative bacteria a pink/ red color.

41. What is the procedure of Gram-staining technique?
a. Take a clean, grease free slide.
b. Prepare the smear of suspension on the clean slide with a loopful of sample.
c. Air-dry or heat-fix smear of cells for around 1 minute with crystal violet staining reagent. (Note: Too heavy or too light cell concentration will affect the Gram Stain results.)
d. Rinse the slide with gentle stream of tap water for 2-3 seconds.
e. Flood the gram’s iodine for 1 minute and wash with gentle stream using tap water for 2-3 seconds.
f. Flood slide with 95% alcohol or acetone for about 15-20 seconds until decolorizing agent running from the slide shows clear liquid.
g. Add counterstain, safranin and wait for 1 minute
h. Wash slide in a gentile stream of tap water until it appear colorless.
i. Blot dry with absorbent paper.
j. Observe under oil immersion using a Brightfield microscope.
k. Results:
Gram-negative bacteria will stain pink or red
Gram-positive bacteria will stain blue or purple
42. What is the procedure of Gram-staining technique?
a. Primary Stain: Crystal Violet Staining Reagent.
Solution A for crystal violet staining reagent
Crystal violet (90% dye content) – 1g
2g Ethanol, 95% (v/v) – 10 ml
Solution B for crystal violet staining reagent
Ammonium oxalate, 0.4 g
Distilled water, 40 ml
Mix solution A and solution B to obtain crystal violet staining reagent. Store for 1 day and then filter using filter paper.
b. Mordant: Gram’s Iodine
Iodine, 0.5 g
Potassium iodide, 1.0 g
Distilled water, 150 ml
Triturate iodine and potassium iodide in a mortar and add water slowly with continuous trituration until the iodine is dissolved. Store in amber bottles.
c. Decolorizing Agent
Ethanol, 95% (vol/vol)
Alternate decolorizing agent – 1:1 acetone and ethanol mixture.
d. Counterstain: Safranin
Use 1.25g Safranin O and mix it with 50 ml 95% Ethanol (Solution A). Take this Solution A 5 ml and mix it with 45 ml distilled water
43. Which indicators are used for spore staining?
Malachite green (a triarylmethane dye) and a safranin (an azonium compound) counterstain is useful tool in identifying the presence or absence of spores.
This is referred to as the Schaeffer-Fulton stain.
44. Explain the Schaeffer-Fulton staining method for endospore staining technique?
1. Prepare a bacterial smear on a clean slide, air dry and gently heat fix.
2. Cover the slide with a piece of paper towel, and place on a staining rack over the water bath with boiling water.
3. Flood the paper towel on the slide with Malachite green (primary stain). Steam the slide for about 5 minutes.
4. Remove the slide from the water bath, and remove the paper towel from the slide.
5. Allow the slide to cool, and then rinse with deionized water until the water runs clear.
6. Drain excess water and apply Safranin (counterstain) for 2 minutes.
7. Rinse Safranin off with deionized water, and blot the slide dry with blotting paper.
8. Examine the slide with a light microscope under oil immersion objective
Result:
Endospores appears green
Vegetative cells appear red or pink

45. What is Lactophenol cotton blue stain or fungal staining?
Lactophenol cotton blue stain is used to examining yeast and filamentous fungi under microscope.
Phenol acts as a disinfectant to kill living organisms
Lactic acid used to preserve the fungal structures
Cotton blue is used to stain fungal cell wall and other fungal structures
On staining, it fungus will be with blue colored spores and structures, such as hyphae.
The identification of fungi using macroscopic and microscopic techniques is difficult and requires a trained eye.


46. What reagents used to prepare Lactophenol cotton blue staining solution?
Distilled water
Cotton Blue or Aniline Blue
Phenol Crystals
Glycerol
Lactic acid
70% ethanol
47. What is use of Ziehl-Neelsen stain?
The objective of Ziehl-Neelsen stain is to differentiate bacteria into acid fast group and non-acid fast groups.
48. What Acid-Fast stain called as Ziehl-Neelsen stain?
This technique was first developed by Ziehl and later on modified by Neelsen, therefore, this method is also called as Ziehl-Neelsen staining techniques.
| Reagents | Color of Acid fast | Color of Non-acid fast |
| Primary dye – Carbol fuchsin | Red | Red |
| Decolorizer – Acid alcohol | Red | Colorless |
| Counter stain – Methylene blue | Red | Blue |

49. What is Total viable aerobic count?
Total viable aerobic count is designed to count the number of microorganisms (as colony forming units, CFUs) in a non-sterile pharmaceutical product or raw materials.
It is father divided into two parts, total aerobic microbial count (TAMC) and total yeast and mould counts (TYMCs).
50. Which are the methods used for analysis of total viable aerobic count?
● Membrane filtration technique – Sample is filtered and the filter is placed on defined media
● Pour plate technique – Sample aliquot is taken and placed in a Petri dish and specified media poured onto the sample
● Spread plate technique – Sample aliquot is placed on the surface of defined media and smeared evenly over the surface
● Most probable number (MPN) technique – This method is used for mainly insoluble materials. The sample dilutions are placed into a series of replicate tubes and the number of tubes showing growth give a statistical evaluation of the number of microorganisms in the sample.
51. Which is Bioburden test Method Validation?
The Bioburden Method Validation is done to demonstrate the adequacy of sample preparation method and the ability of the media to recover microorganisms in the presence of the test sample. Following should be considered during the method validation.
(a) Growth promotion of media
(b) Sample preparation
(c) Test method (d) Sample neutralization
52. Which are the various organization from where approved culture collections of different train of microorganisms can be obtained?
● American Type Culture Collection (ATCC)
● National Collection of Industrial and Marine Bacteria (NCIMB)
● Collection of Institute Pasteur (CIP)
● Imperial Mycological Institute (IMI)
● National Collection of Pathogenic Fungi (NCPF)
● National Biologicals Resources Centre (NBRC)
53. Explain the sample preparation process for Bioburden test.
● Sample handing area: Sample should be prepared on a laboratory bench, within unidirectional airflow cabinet or an isolator.
● Bacteria cultures handling area: Biosafety cabinet or Microbiological Safety Cabinet should be used.
● Sample preparation for Water-soluble products:
a. Dissolve or dilute the product with ration of 1 in 10 dilution in phosphate buffer solution pH 7.2, If necessary, adjust to a pH of 6 – 8.
b. If required, further dilutions can be done to get not more than 250 CFU/plate in case of TAMC, 50 CFU/plate in case of TYMC.
c. If needed, to dissolve the sample completely, triturate it in a sterile mortar and pestle in an aseptic environment to get a fine powder.
● Sample preparation for Non-fatty and insoluble material in water:
a. Suspend the product with 1 in 10 dilution in phosphate buffer solution pH 7.2.
b. A surfactant such as 1 g/L of polysorbate 80 can be used to assist the suspension of poorly wettable substances.
c. If require, adjust to a pH of 6–8.
d. If required, further dilutions can be done to get not more than 250 CFU/plate in case of TAMC, 50 CFU/plate in case of TYMC.
● Sample preparation for fatty products:
a Dissolve in isopropyl myristate sterilized by filtration, or mix the product to be examined with the minimum necessary quantity of sterile polysorbate 80.
b. Heat if required for NMT 40 °C or, in exceptional cases, to not more than 45 °C and maintain the temperature in a water bath.
c. Add pre-warmed diluent to make a 1 in 10 dilution of the product.
d. Form of an emulsion.
c. If needed, further serial tenfold dilution can be prepared using the diluent containing a suitable concentration of sterile polysorbate 80 or another non-inhibitory sterile surfactant.
54. Explain the method of measurement of microbial concentration in suspension by optical density?
A spectrophotometer method is used to measures turbidity of inoculum suspension. When light passes through a suspension of microorganisms, light gets scattered and the amount of scatter is an indication of the concentration present in the suspension.
While doing the estimation using this method, a calibration curve needs to be constructed using series of the known concentration. Based on the calibration curve, unknown concentration can be easily identified.
55. What are the common sources of pyrogens?
Non-bacterial source:
● Antigens
● Poly nucleotides
● Steroids
● Adjuvants
● Viruses
● Fungi
Bacterial source:
● Streptococcal toxins
● Staphylococcal enterotoxins
● Mycobacterial cell wall components
● Bacterial cell wall – lipopolysaccharides (endotoxins)
56. What are the method used to test pyrogens and endotoxins?
● Rabbit pyrogen test
● Limulus Amoebocyte Lysate (LAL) testing for bacterial endotoxin
57. Is sterility test qualitative or quantitative test?
The sterility test is a qualitative test. Results are written as presence or absence of turbidity based on growth for bacteria and fungi in media.
58. Which are commonly used sterility testing methods?
● Membrane filtration: This is further classified as open method and closed system method.
● Direct inoculation method
59. What components required performing sterility testing by membrane filtration method?
● Filter with pore size 0.45 μm
● Filter diameter about 50 mm
● Cellulose nitrate filters for aqueous, oily, and weak alcoholic solutions
● Cellulose acetate filters for strong alcoholic solutions
60. What are the growth mediums used to perform sterility testing?
● Fluid Thioglycollate Medium for anaerobic bacteria and it also isolates aerobic bacteria
● Soya-bean Casein Digest Medium for the isolation of fungi and aerobic bacteria
61. What types of growth promotion test (GPT) to be done for growth medium used to perform sterility testing?
● GPT for Fluid thioglycollate medium shall be done using Clostridium sporogenes, P. aeruginosa, S. aureus;
● GPT for Soya-bean casein digest medium shall be done using A. niger, B. subtilis, C. albicans.
The medium shall be inoculated with not more than 100 CFU and incubated for 3 days (bacteria) or 5 days (fungi).
Result: Clearly visible growth must be observed.
62 What should be incubation conditions and times for sterility testing?
Incubation conditions and times for sterility testing are as follows:
● Fluid Thioglycollate Medium at 30 – 35 °C
● Soya-bean Casein Digest Medium at 20 – 25 °C ● Total incubation period of 14 days with visual examination for turbidity.
63. How to interpret sterility test validation outcome?
There are two outcomes with the sterility test:
(1) Clearly visible growth – test sample and the control tubes are equivalent
(2) No clearly visible comparable growth – Test needs to be modified and the validation needs to be repeated
64. What should be the environment for sterility testing?
The sterility test environment should be EU GMP Grade A with a Grade B background or a Grade A isolator operator with background Grade C or D.
65. How to collect sterility testing sample during the batch?
The test samples for sterility testing needs to be representative of the batch and entire filling operation that is, beginning, middle, and end of the aseptic fill process. Additionally, sterility test sample required to be collected when unplanned intervention occurred during the batch.
66. What is the composition of Fluid thioglycollate medium?
The composition of Fluid thioglycollate medium is as follows:
| Name of ingredient | Weight |
| L-Cystine | 0.5 g |
| Agar | 0.75 g |
| Sodium chloride | 2.5 g |
| Glucose monohydrate/anhydrous | 5.5/5.0 g |
| Yeast extract (water-soluble) | 5.0 g |
| Pancreatic digest of casein | 15.0 g |
| Sodium thioglycollate or | 0.5 g |
| Thioglycollic acid | 0.3 ml |
| Resazurin sodium solution (1 g/l of resazurin sodium), freshly prepared | 1.0 ml |
| Water R | 1000 ml |
pH after sterilization 6.9 to 7.3.
67. What is the composition of Soya-bean casein digest medium?
The composition of Soya-bean casein digest medium is as follows:
| Name of ingredient | Weight |
| Pancreatic digest of casein | 17.0 g |
| Papaic digest of soya-bean meal | 3.0 g |
| Sodium chloride | 5.0 g |
| Dipotassium hydrogen phosphate | 2.5 g |
| Glucose monohydrate/anhydrous | 2.5/2.3 g |
| Water R | 1 000 ml |
pH after sterilization 7.1 to 7.5.
68. While sterility testing using membrane filtration, what should be suitable volume of diluent and sample quantity of product to be examined prescribed?
Liquids sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 1 ml | The whole contents of each container |
| • 1-40 ml | Half the contents of each container but not less than 1 ml |
| • greater than 40 ml and not greater than 100 ml | 20 ml |
| • greater than 100 ml | 10 per cent of the contents of the container but not less than 20 ml |
| Antibiotic liquids | 1 ml |
Insoluble preparations, creams and ointments sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| Insoluble preparations, creams and ointments to be suspended or emulsified | Use the contents of each container to provide not less than 200 mg |
Solids sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 50 mg | The whole contents of each container |
| • 50 mg or more but less than 300 mg | Half the contents of each container but not less than 50 mg |
| • 300 mg – 5 g | 150 mg |
| • greater than 5 g | 500 mg |
Reference: WHO, Document QAS/11.413 FINAL, March 2012
69. Types of filter preferable for sterility testing using filtration method.
Cellulose nitrate filters: For aqueous, oily and weakly alcoholic solutions
Cellulose acetate filters: Strongly alcoholic solutions
70. What is preferred filter diameter for sterility testing using filtration method?
50 mm in diameter
71. Explain Membrane filtration method for sterility testing?
● The filtration apparatus and membrane are sterilized by appropriate means.
● Use membrane filters with nominal pore size not greater than 0.45 μm and diameter 50 mm.
● Solution to be examined shall be filtered under aseptic conditions.
● Aseptically remove the membrane and transfer to the medium.
Method for aqueous solutions:
● Transfer a small quantity of a suitable, sterile diluent such as a 1 g/l neutral solution of meat or casein peptone pH 6.9 to 7.3 onto the membrane in the apparatus and filter.
● Use diluent containing suitable neutralizing substances and/or inactivating substances in the case of antibiotics.
● Transfer the contents of the container or containers to be tested to the membrane or membranes, if necessary after diluting to the volume used in the method suitability test with the chosen sterile diluent but in any case using not less than the quantities of the product to be examined prescribed in Table-1.
● Filter immediately.
● If the product has antimicrobial properties, wash the membrane not less than three times by filtering through it each time the volume of the chosen sterile diluent used in the method suitability test.
● Do not exceed a washing cycle of five times 100 ml per filter, even if during method suitability it has been demonstrated that such a cycle does not fully eliminate the antimicrobial activity.
● Transfer the whole membrane to the culture medium or cut it aseptically into two equal parts and transfer one half to each of two suitable media.
● Use the same volume of each medium as in the method suitability test.
● Alternatively, transfer the medium onto the membrane in the apparatus. Incubate the media for not less than 14 days.
Soluble solids:
Use for each medium not less than the quantity prescribed in Table 3 of the product dissolved in a suitable solvent such as the solvent provided with the preparation, water for injections R, sodium chloride (9 g/l) TS or peptone (1 g/l) TS1 and proceed with the test as described above for aqueous solutions using a membrane appropriate to the chosen solvent.
Oils and oily solutions:
● Use for each medium not less than the quantity of the product prescribed in Table 2.
● Oils and oily solutions of sufficiently low viscosity may be filtered without dilution through a dry membrane.
● Viscous oils may be diluted as necessary with a suitable sterile diluent such as isopropyl myristate R shown not to have antimicrobial activity in the conditions of the test.
● Allow the oil to penetrate the membrane by its own weight then filter, applying the pressure or suction gradually.
● Wash the membrane at least three times by filtering through it each time about 100 ml of a suitable sterile solution such as peptone (1 g/l) TS1 containing a suitable emulsifying agent at a concentration shown to be appropriate in the method suitability test, for example polysorbate 80 at a concentration of 10 g/l.
● Transfer the membrane or membranes to the culture medium or media or vice versa as described above for aqueous solutions, and incubate at the same temperatures and for the same times.
Ointments and creams:
● Use for each medium not less than the quantities of the product prescribed in Table 2.
● Ointments in a fatty base and emulsions of the water-in-oil type may be diluted to 1 per cent in isopropyl myristate R as described above, by heating, if necessary, to not more than 40 °C.
● In exceptional cases it may be necessary to heat to not more than 44 °C.
● Filter as rapidly as possible and proceed as described above for oils and oily solutions.
Table: 1 – Liquids sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 1 ml | The whole contents of each container |
| • 1-40 ml | Half the contents of each container but not less than 1 ml |
| • greater than 40 ml and not greater than 100 ml | 20 ml |
| • greater than 100 ml | 10 per cent of the contents of the container but not less than 20 ml |
| Antibiotic liquids | 1 ml |
Table: 2 – Insoluble preparations, creams and ointments sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| Insoluble preparations, creams and ointments to be suspended or emulsified | Use the contents of each container to provide not less than 200 mg |
Table: 3 – Solids sample
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 50 mg | The whole contents of each container |
| • 50 mg or more but less than 300 mg | Half the contents of each container but not less than 50 mg |
| • 300 mg – 5 g | 150 mg |
| • greater than 5 g | 500 mg |
Reference: WHO, Document QAS/11.413 FINAL, March 2012
72. Explain the direct inoculation of the culture medium for sterility testing?
● Transfer the quantity of the preparation to be examined prescribed in the following Table directly into the culture medium so that the volume of the product is not more than 10% of the volume of the medium, unless otherwise prescribed.
Table: 1 – Liquids samples
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 1 ml | The whole contents of each container |
| • 1-40 ml | Half the contents of each container but not less than 1 ml |
| • greater than 40 ml and not greater than 100 ml | 20 ml |
| • greater than 100 ml | 10 per cent of the contents of the container but not less than 20 ml |
| Antibiotic liquids | 1 ml |
Table: 2 – Insoluble preparations, creams and ointments samples
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| Insoluble preparations, creams and ointments to be suspended or emulsified | Use the contents of each container to provide not less than 200 mg |
Table: 3 – Solids samples
| Minimum quantity to be used for each medium Quantity per container | Minimum quantity to be used for each medium unless otherwise justified and authorized |
| • less than 50 mg | The whole contents of each container |
| • 50 mg or more but less than 300 mg | Half the contents of each container but not less than 50 mg |
| • 300 mg – 5 g | 150 mg |
| • greater than 5 g | 500 mg |
● If the product to be examined has antimicrobial activity, carry out the test after neutralizing this with a suitable neutralizing substance or by dilution in a sufficient quantity of culture medium.
● When it is necessary to use a large volume of the product it may be preferable to use a concentrated culture medium prepared in such a way that it takes account of the subsequent dilution.
● Where appropriate the concentrated medium may be added directly to the product in its container.
Oily liquids:
● Use media to which have been added a suitable emulsifying agent at a concentration shown to be appropriate in the method suitability of the test, for example polysorbate 80 at a concentration of 10 g/l.
Ointments and creams:
● Prepare by diluting to about 1 in 10 by emulsifying with the chosen emulsifying agent in a suitable sterile diluent such as peptone (1 g/l) TS1.
● Transfer the diluted product to a medium not containing an emulsifying agent.
● Incubate the inoculated media for not less than 14 days. Observe the cultures several times during the incubation period.
● Shake cultures containing oily products gently each day. However when fluid thioglycollate medium is used for the detection of anaerobic microorganisms keep shaking or mixing to a minimum in order to maintain anaerobic conditions.Reference: WHO, Document QAS/11.413 FINAL, March 2012
Most useful 80+ HPLC Interview Questions and troubleshooting

HPLC interview questions are the most commonly asked topic during the quality control laboratory interview for the pharmaceutical industry. HPLC is the pharmaceutical industry’s most widely used technique of analysis; therefore, most of the interview questions and answers are around HPLC technique and HPLC troubleshooting.
You will find interview questions and answers on components of an HPLC system, isocratic and a gradient HPLC system, types of gradient mixing, function of the HPLC column, types of sample injector, function of the HPLC column oven, commonly used detector in HPLC systems, types of detectors used in HPLC systems, typical HPLC startup procedure, procedure to flush the new reversed-phase column, checkpoints before starting an HPLC analysis, relative retention time (RRT) in liquid chromatography, ‘fronting’ or ‘leading’ in liquid chromatography, peak symmetry, signal-to-noise ratio, procedures for mobile phase degassing, advantages of acetonitrile in the mobile phase, advantages of methanol in the mobile phase, reasons for variable Retention Times (RT) in HPLC analysis, and many other relevant and valuable HPLC interview questions and answers.
The interview questions cover questions from basic to advanced levels of technical aspects. These HPLC interview questions and answers will help crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning and best of luck with your interview! Happy Learning.
Following are most useful HPLC Interview Questions and answers as well as HPLC troubleshooting guide
1. What are the essential components of an HPLC system?
HPLC instrument consists:
- An eluent delivery system (= pump)
- An injector, a column
- A detector and a data evaluation system
2. What is the difference between an isocratic and a gradient system?
It can be distinguished easily. If there is only one inlet tube for the eluent, it is an isocratic instrument, and, there are two or more are present, its a gradient system.
3. What is the gradient HPLC system?
In a gradient system, two or more solvents are continuously mixed during the separation.
4. What are the types of gradient mixing?
(a) When mixing of solvent happens before the pump by a proportional valve, it is a low pressure gradient.
In a low pressure gradient system, the mixing happens in the normal pressure or low pressure side of the device before the pump.
(b) When the mixing happens after the pump on the high pressure side, mixing takes place in a mixing chamber where the solvents of both pumps meet. Such an instrument has a high pressure gradient.
5. What are the types of sample injector?
(a) Sample introduction with a hand injector or a manual valve
(b) Sample introduction with an autosampler
6. What is the function of the HPLC column?
The HPLC column is the heart of the system. The function of the HPLC column is to separate the compound with various separation mechanisms.
7. What is the function of the HPLC column oven?
Function of the column oven is to maintain a constant temperature of the column and support to get reproducible results.
8. What is the most commonly used detector in HPLC systems?
UV detector and diode array detector (also called as PDA – Photodiode Array).
9. What are the various types of detectors used in HPLC systems?
(i) UV-Visible HPLC Detector
(ii) PDA Detectors (Diode Array Detector or Photo Array Diode Detector) HPLC detector
(iii) Refractive-Index HPLC Detector
(iv) Evaporative Light Scattering Detector (ELSD) HPLC Detector:
(v) Multi-Angle Light Scattering Detector (MALS) for HPLC:
(vi) HPLC-Mass Spectrometer (HPLC-MS) Detector
(vii) HPLC Conductivity Detector:
(viii) HPLC-Fluorescence Detector:
(ix) Chemiluminescence HPLC Detector:
(x) HPLC Optical Rotation Detector or Chiral Detector
(xi) Electrochemical (Amperometric) Detector for HPLC
(xii) HPLC Photoconductivity Detectors
(xiii) HPLC Infrared (IR) Detectors
(xiv) Laser-Induced Fluorescence Detector
(xv) Radioactivity Detector
(xvi) HPLC-NMR Detector
10. What is used to transfer the mobile phase from one module to another?
The mobile phase is transferred from one module to the another module through capillaries made up of stainless steel or PEEK (polyetheretherketone).
11. What is the internal diameter of the HPLC capillaries?
The typical internal diameter of the HPLC capillaries between pump and injector is 0.5-1 mm. The typical internal diameter of the HPLC capillaries after the outlet of the injector is less than 0.2 mm.
12. What is the internal diameter of the HPLC capillaries when back pressure is required to achieve?
Detectors such as fluorescence detectors need some back pressure for adequate operation which can be achieved with a 0.1-0.2 mm capillary internal diameter.
13. What are restrictor capillaries, sometimes also simply called or restrictors?
A restriction capillary or restrictors are the capillaries with very narrow internal diameter, that restrict the mobile phase flow. It has the capability to closely mimic the normal operating conditions by generating back-pressure of 1k to 2k psi.
14. What is the reason for using interconnection pieces of HPLC from the same manufacturer?
Interconnection pieces of HPLC such as ferrules and fittings should be used from the same manufacturer, because while using different make, they may have different dimensions and internal diameters leading to a small dead volume. This can cause abnormality during separation..
15. What is the typical HPLC startup procedure?
a. Flush system without column at a flow rate of 1 ml/min with a 50/ 50 mixture isopropanol/ water for about 10 min.
b. Inject the mobile phase a few times in order to ensure that the old eluent or impurities are removed from the sample injection system.
16. What precautions should be considered before the first sample run in the HPLC?
i. Once the mobile phase is prepared and run in the system, leave the HPLC system for a little time to equilibrate to flush impurities or dirt if any out of the column.
ii. Dissolve samples adequately as per method of analysis.
iii. No particles should remain in the sample, use membrane filtration as per method.
iv. Ensure that your dissolved sample should not precipitate in the mobile phase.
v. Inject a standard and take a look at the chromatogram.
vi. Ensure that the baseline is stable with no drift and peaks are symmetrical.
vii. Ensure that the chromatogram after the second injection is identical to the first one.
17. How to flush the new reversed-phase column?
While installing a new reversed-phase column, flush it with acetonitrile or methanol before use it for the first run.
18. How to ensure that system is free of buffers?
Flush the system first with an isopropanol/ water mixture and then with methanol.
19. What is the procedure to temporarily stop the HPLC (When you are aware that you will run the HPLC on the next day or after a pause)?
When you know that you will run the HPLC (same setup) after some time, shut down all components of the instrument except the pump. Keep the pump running at a lower flow rate about 0.1-0.3 ml/min.
Ensure that sufficient mobile phase is available. Do not run HPLC dry. While resuming the HPLC again, adjust the flow as per the method.
20. What is the procedure to stop the HPLC?
When you want to stop HPLC for a longer duration, flush the system using water to remove the buffer out from the system. Followed by flushing the HPLC using 20-30 ml methanol or acetonitrile.
Store the column using acetonitrile or methanol. Close the column with end fittings to prevent drying of the stationary phase.
21. What is the preferred solvent to store the column if the column need not be used for a longer time? Why?
Acetonitrile is a preferred solvent as it is stable for a longer period of time compared to methanol. Methanol has the property of hydrolysis.
22 What are the checkpoints before starting an HPLC analysis?
1. Ensure no electrical connection is loose.
2. Verify the capillaries for absence of leakages.
3. Check the tubing and solvent container for absence of air bubbles. If any air bubbles are observed, remove air from tubes by sucking solvent with the help of a syringe by opening the purge valve. Perform the purging and priming of the system.
4. Ensure that the solvent container is closed to prevent objects falling into it as well as evaporation of solvents.
5. Check the system flow by switching the pump and ensure that solvent is coming from mobile phase container/ solvent containers to the waste container.
6. Probable reasons of absence of flow are:
a. Air in pump
b. Verify the leakage by touching the seals. It should not be wet.
c. Crystals of buffered mobile phases.
d. Malfunctioning can be verified through unusual pump noise
7. Ensure that mobile phase is prepared using HPLC grade solvents
8. Ensure that the mobile phase is prepared by counting required number of injections, run time and flow rate.
9. Ensure that buffered mobile phases are filtered using a membrane filter, degas with helium or with degasser.
10. While using a manual injector ensure that the container is kept under the overflow.
11. Maintain the injection needles clean to avoid contamination when using manual injector.
12. Clean injector needle using suitable solvent if needed.
13. Use a washing solution while using an autosampler.
14. Check the UV detectors lamp energy when using the UV detector.
15. For RP-HPLC, use 10-20% methanol in the water to prevent microorganism growth.
16. Use a waste container to collect waste. Ensure the safety precautions to prevent spillage and evaporation in the lab.
17. Before starting the standard, verify the baseline.
18. Before starting the sample, verify the peak shape of standard, system suitability and area.
23. What is the meaning of Peak in chromatography?
Peak is a detector response represented as a chromatogram. One peak represents one component when separation is done adequately. When there is incomplete separation, i.e. two or more components’ peak gets merged because they eluted in overlapping manner and remain unresolved.
24. What is a Chromatogram?
Chromatogram a representation of detector response or a quantitative measurement value in a graphical form vs. the volume or time. Typical chromatograms have series of Gaussian peaks on a baseline.
25. What is Retention time (tR) in liquid chromatography?
Retention time, (tR) is the time between the injection of the compound and the maximum peak response of the eluted.
26. What is Dwell volume (D) in chromatography?
The volume between the is the volume between the point at which the eluents (mobile phase/ solvent) meet and the inlet of the column (Or the top of the column).
Dwell volume is also known as “gradient delay volume”.
27.What is Hold-up time (tM) in chromatography?
The time required for elution of an unretained component (In following schematic, an air or unretained solvent peak, with the baseline scale in minutes).

Reference: Working document QAS/21.905 – February 2022 (draft), 1.14.1 CHROMATOGRAPHY
28. What is Hold-up volume (VM) in chromatography?
The volume of mobile phase or solvent required for elution of an unretained component. It is
calculated using hold-up time ((tM)) and the flow rate (F), mL/min.
Formula is VM = tM × F
29. What is Number of theoretical plates (N) OR Plate number (N) in chromatography?
Number of theoretical plates (N) OR Plate number (N) is a measure that indicates column efficiency. In other words, it is a number that is indicative of performance of column OR column efficiency.
For Gaussian peaks, it is calculated by:
N = 16(tR/W)^2 (As per USP)
tR – retention time
W – peak width
For electronic integrators it can be determined using the equation:
N = 5.54 (tR/Wh)^2
(As per USP and Working document QAS/21.905 – February 2022 (draft), 1.14.1 CHROMATOGRAPHY)
tR – retention time
Wh – peak width at half-height (h/2).
30. What is Plate Height (H) in chromatography?
Theoretical plate height is height equivalent to a theoretical plate. It is a ratio of the column length (L), in micrometers, to the plate number (N): H = 𝐿/𝑁
It indicates the rate for the band broadening of peak broadening in HPLC equipment. The smaller the H value, the bigger the plate number.
When bigger the plate number, the better the column is packed and the smaller the dead volume of the instrument and, the sharper the peaks. It also means that the efficiency of the column is good.
31. What are the factors affecting the Number of theoretical plates (N) or Plate Height (H)?
- Substance being chromatographed
- Operating conditions i.e flow rate and temperature of the mobile phase or carrier gas
- Quality of the packing material
- The uniformity of the packing within the column
- For capillary columns, the thickness of the stationary phase film
- Internal diameter and length of the column
32. What is the meaning of Resolution (RS) in chromatography?
The resolution is the separation of two components in a mixture.
It is calculated using following formula:
RS = 2 × (tR2 − tR1)/(W1 + W2)
tR1 and tR2 are the retention times of the two components
W1 and W2 are the corresponding widths at the bases of the peaks obtained by extrapolating the relatively straight sides of the peaks to the baseline.
For electronic integrators it is determine using following formula:
RS = 1.18 × (tR2 − tR1)/(W1,h/2 + W2,h/2)
33. What is the meaning of Retention volume (VR) in chromatography?
Retention volume (VR) a volume of mobile phase or solvent required for elution of a component.
It is calculated using formula VR = tR (retention time) × F (flow rate in mL/min)
34. What is Peak-to-valley ratio (p/v) in liquid chromatography?
The peak-to-valley ratio can be considered as a system suitability criterion for related substances test in the condition of non achieving separation of two peak at the baseline level. Schematic representation is given as follows.


35. What is Relative retention (r) in liquid chromatography?
Relative retention (r) is the ratio between the adjusted retention time of a component relative to that of another used as a reference obtained under same chromatographic conditions.
It can be calculated using formula r = (tR2 − tM)/(tR1 − tM)
tR2 – retention time measured from the point of injection of the compound of interest
tR1 is the retention time measured from the point of injection of the compound used as reference
tM is the retention time of a non retained marker defined in the procedure
36. What is Relative retention time (RRT) in liquid chromatography?
Relative retention time (RRT) is also known as the “unadjusted relative retention”.
It is calculate using formula RRT = tR2/tR1
37. What is “Symmetry Factor” or “Tailing Factor” in liquid chromatography?
The Gaussian form of peak is generally completely symmetrical. When the rear is spread out to more or less extent and forms a ‘tail’. This phenomenon is called ‘tailing’.
“Symmetry Factor” or “Tailing Factor” (AS) is calculated using formula: W0.05/2f
W0.05- width of the peak at 5% height
f is the distance from the peak maximum to the leading edge of the peak
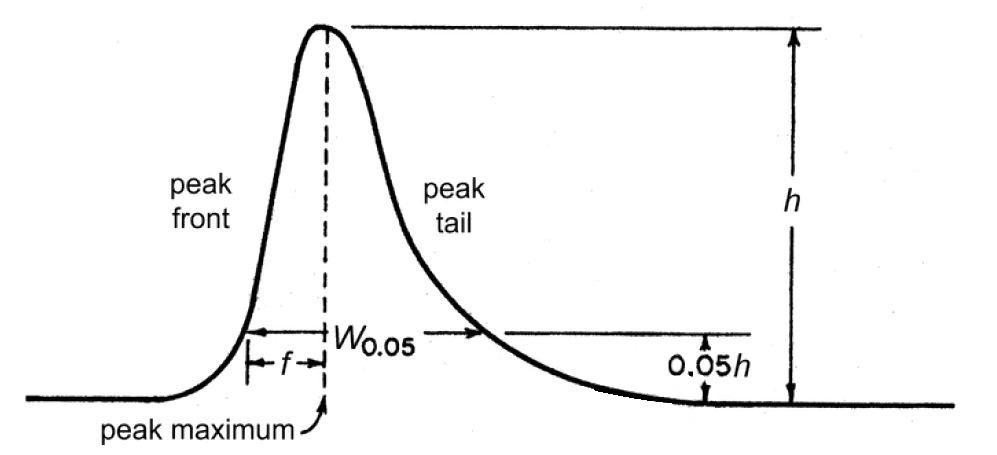
Reference: USP
38. What is ‘fronting’ or ‘leading’ in liquid chromatography?
In opposite of tailing phenomenon; when the front is flatter than the back, it is called as
‘fronting’ or ‘leading’.

39. What is System repeatability in liquid chromatography? OR What is System Suitability?
The repeatability and reproducibility of signal/ area/ response that is represented as % Relative Standard Deviation (% RSD) of consecutive measurements for Not Less Than (NLT) three standard solutions or applications of an applicable reference solution.
Total mobile phase time (tt) 279
In size-exclusion chromatography, the retention time of a component whose molecules 280 are smaller than the smallest gel pores (Figure 5).
40. What is the total mobile phase volume (Vt)?
This phenomenon is applicable for size-exclusion chromatography. The retention volume of a compound for those molecules having smaller than the minimum gel pores.
It may be measured using the flow rate (F) in ml/min and total mobile phase time. The equation is 𝑉𝑡=𝑡𝑡×𝐹
41. What is an ideal criteria for peak symmetry?
If not specified in the monograph or test method such as assay, related substance or other test, the symmetry factor or tailing factor applicable for the peak during quantitative evaluation is 0.8 to 1.8.
42. What is a signal-to-noise ratio?
The signal to noise ratio represents the capability of a method to detect or quantify the elute in a consistent manner; in other word, the signal-to-noise ratio represents the system sensitivity.
When the signal-to-noise ratio is more than 3, compounds can be consistently detected for each time. When the signal-to-noise ratio is more than 10, compounds can be consistently quantified for each time
43. What are the issues if the Reverse Phase HPLC column (stationary phase) is not cleaned properly?
Inadequate cleaning of the Reverse Phase HPLC column (stationary phase) results in a high back pressure, decrease in separation performance, broadening of peak, peak tailing and sometimes “ghost peaks” are detected.
This happens because of hydrophobic organic molecules, for example, lipids or large organic molecules easily stuck to RP HPLC Columns.
44. What is the procedure to clean the Reverse Phase HPLC column (stationary phase) quickly and efficiently?
Inject 100 to 200 microliter methanol or acetonitrile. Repeat this procedure 2 to 3 times. If a garbage peak still observes, do the normal flush with methanol or acetonitrile.
If you are using acetonitrile, ensure that it does not cause a precipitation of buffer-containing eluents.
45. What happens when the mobile phase is not degassed?
Noisy or drifting baselines and pressure fluctuations are signs of insufficient degassing.
46. What are the various procedures used for mobile phase degassing?
• Refluxing
• Vacuum degassing
• Helium degassing
• Ultrasonic degassing
47. What are the suggested procedures for mobile phase degassing?
• Refluxing – Method is good but it is not practicable.
• Vacuum degassing – Method is good.
• Helium degassing – Method is good.
• Ultrasonic degassing – Ineffective method. Works good for acetonitrile/ water mixtures.
48. What are the advantages of acetonitrile in the mobile phase?
- Low viscosity and better kinetics provides sharper peaks.
- acetonitrile/water mixtures have lower back pressure in comparison to methanol/water mixtures, hence, less wear and tear on seals and columns.
- Higher elution strength, henace, lower solvent consumption. It provided similar elution strength at a lower concentration compared to methanol.
- Silica gel is less prone to dissolve in acetonitrile compared to methanol (Reason: acetonitrile is less polar than methanol)
- Low solubility of air, hence, less problems with air and easy for effective degassing.
- Low UV absorptivity hence, better for detection at 195-200 nm
- Better reproducibility, specifically while using ionic solutes because it causes small pH deviations in aqueous solutions.
- Suitable for ion chromatography because it is a better solvating agent.
- More toxic than methanol resulting in prevention of microbiological growth in the instrument.
- Suitable for the separation of bases at lower pH and yield sharp peaks
49. What are the advantages of methanol in the mobile phase?
- Methanol is odorless hence, it provides better working conditions.
- Less toxic compared to acetonitrile.
- Better solubility for salts hence, chances of precipitation low.
- Methanol/water mixture brings the seals faster into its swelling conditions hence, equipment gets faster to working condition
- On aging of acetonitrile, impurities such as propionitrile, methacrylonitrile generate ghost'” peaks. This issue is less known for methanol hence, longer shelf life of methanol.
- Good separation of bases at alkaline pH
- Lower baseline noise above 220 to 230 nm
50. What could be the impact of too high or too low pH of the mobile phase on the C18 column?
Impact of high pH environment on C18 Column is silica gel dissolves at above pH 8. Because of this, the column performance will rapidly decrease.
Impact of low pH environment on C18 Column, C18 chains are hydrolyzed at pH 2 and lower. Because of that, the column bleeds, and the performance decreases.
51. What is the solution to deal with very high (above pH 8) or very low pH (at pH 2 and lower) mobile phase?
Install a pre-column or old CI8 column between the pump and the injector.
On installation of this precalumn, the alkaline eluent will get saturated with silica gel and will not affect the separation column.
At low pH, the C18 chains of the pre column are hydrolyzed, protecting the main column.
52. What are the possible reasons for a change in retention time in HPLC analysis?
Following are potential reasons for change in retention time in HPLC analysis.
Change in stationary phase
Change in mobile phase
Change in temperature
Change in flow rate
Change in packing density
53. What are the most probable reasons for the short life span of the HPLC Columns?
- Strongly adsorbed contaminants in the sample can ruin the column performance
- Shedding seals may clog column filters and the top layers of the packing.
- Mechanical weakness of the packed bed which could be the consequence of rough handling of the column while handling in the lab or during shipment..
- Inconsistent column life may occur because of adsorption of sample constituents on the top of the column. This could either occur because of precipitation because of low solubility of sample in the mobile phase or they may be strongly adsorbed. This can also happen if one injects more samples. In that case, contaminants build up on the top of the column and prevent the sample to properly adsorb and distribute. This issue results in a peak distortion and an increase in back pressure.
- Column collapsing may occur if a mobile phase pH outside the recommended range of column.
- Sample dissolved in a strongly acidic or alkaline solution.
- Columns exposed to the wrong solvents.
- Repeated column “washing”.
54. What are the solutions to increase the life span of the HPLC Columns?
- Sample clean up with a suitable sample preparation method.
- Use a guard column or precolumn. This will prevent column clogging and accumulation of contaminants at the top of the column. To have the highest performance of the guard column, use exactly the same packing as the analytical column.
- (Not preferred method) Column washing could be one of the solutions that dissolves the contaminants on the top of the column. The drawback is washing will remove hydrolyzed bonded phase. Therefore, a repetitive washing may result in accelerated aging of the column.
- (Not preferred method) Column backflushing. The drawback is when washing with a different solvent than the mobile phase, same consequences as column washing. While doing this using the mobile phase, it’s time consuming and sometimes it doesn’t work. (Note: Backflushing should not be made as standard practice).
55. What are the possible reasons for variable Retention Times (RT) in HPLC analysis?
Randomly changing and inconsistent retention times – Pump(s) and the solvent mixing devices malfunction and not giving consistent flow.
- Retention time does not change from run to run but they vary from day to day – most likely the source of variation is the composition of the mobile phase. Note: Rule of thumb, if you make an error of 1% in the amount of organic solvent, the retention time can change by between 5% and 15%, typically by about 10%.
- Temperature fluctuations (Rule of thumb – RT change by about 1% – 2% per 1 ºC).
- Air conditioning shut down usually at weekends.
56. What are the solutions to prevent the variation in retention time in HPLC analysis?
- Accurately preparation of the mobile phase. While preparing the mobile phase, measure the amount of solvent very carefully. Preferred method to prepare the mobile phase gravimetrically rather than volumetrically.
- Method of degassing of mobile phase contributes to variability. Very good degassing method is applying vacuum and ultrasound together for about one minute. Consequently there will be very good degassing with a nominal amount of solvent evaporation. Another good method is the use of helium sparging. Precaution: After initial equilibration, the flow of helium to be reduced to prevent helium to carry solvent vapors with it and reduce evaporation.
- Accurate measurement of pH. A change of as little as 0.1 pH units can result in a retention time shift of 10%.
- Control of the pH is critical when your sample contains ionic or ionizable compounds.
- Temperature is an important factor to prevent the fluctuation of retention time.
57. What are the reasons for Drifting Retention Times in HPLC analysis?
Most common cause of drifting retention times is an equilibration problem.
In normal phase chromatography the Retention time is sensitive to the quantity of water adsorbed on the silica surface. Solubility of water in solvents such as methylene chloride or hexane is very low, column equilibration takes time. Still there are chances that RT my shift when dry hexane is used.
Equilibration in reversed-phase chromatography is quicker; 5 to 10 column volumes of mobile phase is generally sufficient to equilibrate column.
58. What is the solution for prevention of drifting Retention Times in HPLC analysis?
As we understood, the most common cause of drifting retention times is an equilibration problem.
In normal phase chromatography it is recommended to avoid using very dry solvents. Most preferable approach to solve the equilibration problem of silica with water is to use solvents that are “half-saturated” with water. It can be prepared by saturating a provided volume of hydrophobic solvent with water and mixing it 1:1 with “dry” solvent. This will facilitate quick equilibration of the column.
In reversed-phase chromatography, 5 to 10 column volumes of mobile phase are generally good for quick equilibration.
59. What are the possible reasons for the peak shape issue of Peak Tailing?
- Wrong mobile phase pH
- Column void
- Blocked frit
- Unswept dead volume
- Interaction with active silanols
- Chelation with metal ions in stationary phase
60. What are the solutions to prevent peak shape issues of Peak Tailing?
- Wrong mobile phase pH → Increase buffer concentration or Decrease mobile phase pH that suppress silanol ionization
- Column void → Change the column, Column backwash
- Blocked frit → Column backwash, Use inline filters
- Unswept dead volume → Use shorter tubing connection and minimum number of connection, Ensure tightening of all connections
- Interaction with active silanols or Chelation with metal ions in stationary phase → Add basic mobile phase additive or Use ultra-high purity silica based stationary phase
61. What are the possible reasons for the peak shape issue of Split Peaks?
- Contamination on column inlet
- Incompatibility of Sample solvent with mobile phase
- Elution of second component simultaneous
- Blocked frit
- Column overloaded
62. What are the solutions to prevent peak shape issues of Split Peaks?
- Contamination on column inlet → 1. Use guard column, or replace guard column, or replace analytical column (depending on issue identified), 2. Backwash analytical column (less preferred method and should be rarely used), 3. If contaminants are strongly adsorbed, try to regenerate column Or 4. Replace column if issue is not resolved
- Incompatibility of Sample solvent with mobile phase →
- Elution of second component simultaneous → 1. Sample cleanup before injection, 2. Modify mobile phase composition, or 3. Change stationary phase depending on selectivity
- Blocked frit → Use in-line filter, Column backwash
- Column overloaded →1. Increase column capacity by using high capability stationary phase or increase the dimension, or, 2. Decrease injection volume
63. What are the possible reasons for the peak shape issue of Peak Fronting?
- Sample solvent incompatible with mobile phase
- Formation of channels in column
- Low temperature of column oven
- Column overloaded
64. What are the solutions to prevent peak shape issues of Peak Fronting?
- Low temperature of column oven → Increase column oven temperature to increase column temperature
- Column overloaded → Use less injection volume or dilute the sample solution according to desired column load
- Formation of channels in column → Refer to the column literature to operate the system within the given range of parameters, e.g. pH of the solution. If problem persist, replace the column
- Sample diluent incompatible with mobile phase → Change the sample diluent as mobile phase
65. What are the types of Retention Time Variation or RT Variation?
- Decreasing Retention Times
- Increasing Retention Times
- Fluctuating Retention Times
66. What are the probable reasons for Decreasing Retention Times?
- High flow rate
- Column overloaded
- Loss of bonded stationary phase
- Active groups on stationary phase
- Incorrect column selection
67. What are the solutions to prevent the issue of Decreasing Retention Times?
- High flow rate – Check and regulate the flow rate of pump
- Column overloaded – Decrease sample injection volume, Select the column with larger internal diameter
- Loss of bonded stationary phase – Replace column, Operate at recommended pH range for RP columns. Generally operated at 2-8 for silica based RP HPLC.
- Active groups on stationary phase – Increase buffer strength
- Incorrect column selection – Select the column with larger internal diameter
68. What are the probable reasons for Increasing Retention Times?
- Changing mobile phase composition
- Low flow rate
- Loss of bonding in stationary phase
- Bubbles in mobile phase
69. What are the solutions to prevent the issue of Increasing Retention Times?
- Changing mobile phase composition – 1. Cover solvent reservoirs, 2. Prepare fresh mobile phase
- Low flow rate – 1. Check and adjust pump flow rate, 2. Check for leaks in system, including pump seals
- Loss of bonding in stationary phase – Replace column
- Bubbles in mobile phase – 1. Check flow rate and pressure, 2. Degas mobile phase
70. What are the probable reasons for Fluctuating Retention Times?
- Inadequate column equilibration
- Mobile phase preparation error/ composition variation
- Inadequate buffer capacity
- Unstable column temperature
71. What are the solutions to prevent the issue of Fluctuating Retention Times?
- Unstable column temperature – 1. Check column oven to ensure temperature stability, 2. Malfunctioning of the column oven thermostat
- Inadequate column equilibration – 1. Allow columns to equilibrate for sufficient time between runs, 2. Condition the column using concentrated sample
- Mobile phase preparation error/ composition variation – 1. Verify volume make-up of mobile phase, if needed, prepare fresh one, 2. Verify the proportioning-valve accuracy
- Inadequate buffer capacity – Utilize the buffer concentrations more than 20mM
72. What are the probable reasons for Ghost Peaks?
- Contamination of column
- Contamination of injector
- Carryover/ late eluting peak from previous injection
- Contaminated water or solvent
- Method specificity not established adequately
73. What are the solutions to prevent the issue of Ghost Peaks?
- Contamination of column – Adequate column washing/ Flushing of column to remove contaminants
- Contamination of injector – Injector flushing between analyses
- Carryover/ late eluting peak from previous injection – 1. Increase the run time, 2. Flush column with strong mobile phase at end of each run, 3. For gradient runs, end the run at higher concentration
- Contaminated water or solvent – Use HPLC grade water or solvent
- Sample contamination – Ensure cleanliness of glasswares, sample preparation aids such as mortar and pestle, cleanliness of sample storage area
- Method specificity not established adequately – Use sample clean-up process and perform method specificity covering all the relevant factors (excipient peak interference, degradants, etc.)
74. What are the probable reasons for Negative Peaks?
- When using RI detector, Refractive Index of solute is less than the mobile phase
- When using UV detector, solute absorption is less than absorption of mobile phase
- Different composition of Sample solvent and mobile phase
75. What are the solutions to prevent the issue of Negative Peaks?
- When using the RI detector, the Refractive Index of solute is less than the mobile phase – 1. Use mobile phase with less refractive index, 2. Invert detector polarity to get positive peaks
- When using a UV detector, solute absorption is less than absorption of mobile phase – 1. Change UV wavelength to get a positive peak, 2. Identify mobile phase so that it lowers the UV absorption
- Different composition of Sample solvent and mobile phase – Revise the composition of either sample solvent and or mobile phase to improve the compatibility and sample solubility
76. What are the probable reasons for Spikes in a chromatogram?
- Air bubbles in mobile phase/ inadequately degassing of mobile phase
- Column stored without closing ends using column closure caps
77. What are the solutions to prevent the issue of Spikes in a chromatogram?
- Air bubbles in mobile phase/ inadequately degassing of mobile phase – 1. Degas mobile phase using suitable method, 2. Use back pressure restrictor at detector outlet, 3. Ensure all tubings are without any leaks and fittings are tight
- Column stored without closing ends using column closure caps – 1. Ensure that column is stored with end caps closed, 2. Flush Reverse Phase column using degassed methanol
78. What are the probable reasons for High Back Pressure in HPLC systems?
- Wrong HPLC pump setting
- Pressure higher during middle of gradient
- Temperature lower than required
- Column ageing
- Column frit blockage
- In-line filter blockage
- Guard column blockage
- System blockage
- Buffer precipitation in the system
79. What are the solutions to prevent the issue of High Back Pressure in HPLC systems?
- Wrong HPLC pump setting – Verify HPLC pump setting and adjust appropriately
- Pressure higher during middle of gradient – This is normal phenomenon
- Temperature lower than required – Maintain the column oven temperature as per method requirement
- Column ageing – It is normal phenomenon of gradual increase in pressure over the period of lifetime
- Column frit blockage – 1. Column backwash, 2. Use an in-line filter to prevent column blockage and reduce backpressure, 3. Ensure efficient filtration, 4. Use guard columns
- In-line filter blockage – 1. Replace in-line filter frit with new one, 2. Centrifuge or filter samples with recommended filters, 3. Degas and Pre-filter mobile phase
- Guard column blockage – Increase the replacement frequency depending on experience
- Buffer precipitation in the system – Back flush the column using water
80. What are the probable reasons for Low Back Pressure in HPLC systems?
- Leakage in the HPLC system
- Column temperature is higher than required
- Flow rate is lower than required
81. What are the solutions to prevent the issue of Low Back Pressure in HPLC systems?
- Leakage in the HPLC system – Identify the leakage and correct it
- Column temperature is higher than required – Set the temperature as per method requirement
- Flow rate is lower than required – Set the flow rate as per the method requirement
82. What are the good habits that help to minimize the HPLC system related issues such as peak shape problems, retention time variation, ghost peaks and column back pressure issues?
- For new projects, select well researched, high-purity silica-based column and use highest quality HPLC-grade reagents.
- Flush the HPLC system at regular intervals that removes salts and buffers.
- Service the system periodically to reduce check-valve and pump-seal problems.
- Precise sample preparation with adequate filtration and sample clean-up process reduces sample related issues.
- Strong-solvent flush after every run or at specific frequency will reduce sample carryover and extend column lifetimes.
- Columns won’t last forever, but with proper care, you should be able to get a good return on your investment.
Refer Complete guide on High Performance Liquid Chromatography (HPLC)
Useful 280+ Pharmaceutical Quality Control Interview Questions and Answers

Quality Control Interview Questions listed in this article are the most commonly asked topic during the quality control laboratory interview for the pharmaceutical industry and chemical industry. In this article we tried cover pharmaceutical industry’s most widely used technique of analysis.
You will find interview questions and answers on Basics chemistry terminology of concentration calculations, Stability Studies, UV/ Visible spectrophotometry, Fluorescence Spectroscopy, Mid and Near Infrared Spectroscopy, Raman Spectroscopy, Thermal analysis techniques, Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA), Data handling in analytical chemistry, Weighing balance and weighing techniques, Volumetric glasswares, Titrations and standardization, Gas Chromatography (GC), Techniques of pharmaceutical analysis, Errors in pharmaceutical analysis, Significant figures, Pharmacopoeia, Impurities in the pharmaceuticals, Limit tests in the pharmaceuticals, Electrochemical methods of analysis in the pharmaceuticals, Karl fischer method for determination of water, Optical method of analysis, Nuclear Magnetic Resonance (NMR) spectroscopy, Emission spectroscopy, Flame Emission Spectroscopy (FES), Atomic Absorption Spectrophotometer, Thin-Layer Chromatography (TLC), and HPLC Interview Questions and troubleshooting and many other relevant and valuable quality control interview questions and answers.
The interview questions cover questions from basic to advanced levels of technical aspects. These quality control interview questions and answers will help crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning and best of luck with your interview! Happy Learning
Pharmaceutical Quality Control Interview Questions and Answers
Basics chemistry terminology of concentration calculations
1. What is atomic weight for any element?
The atomic weight for any element is the weight of a specified number of atoms of
that element, and that number is the same from one element to another.
2. What is gram atomic weight for any element?
A gram-atomic weight of any element contains exactly the same number of atoms of that element as there are carbon atoms in exactly 12 g of carbon 12.
3. What is Avogadro’s number?
This number is Avogadro’s number, 6.022 × 1023, the number of atoms present in 1 g-at wt of any element.
4. What is molecular weight?
The molecular weight is defined as the sum of the atomic weights of the atoms that make up a compound.
5. What is formula weight OR molar mass?
The term formula weight or molar mass is a description for substances that don’t exist as molecules but exist as ionic compounds (strong electrolytes—acids, bases, salts).
6. What is dalton?
Biologists and biochemists sometimes use the unit dalton (Da) to report masses of large biomolecules and small biological entities such as chromosomes, ribosomes, viruses, and mitochondria, where the term molecular weight would be inappropriate.
The mass of a single carbon-12 atom is equivalent to 12 daltons, and 1 dalton is therefore 1.661 × 10−24 g, the reciprocal of Avogadro’s number. The number of daltons in a single molecule is numerically equivalent to the molecular weight (g/mol).
7. What is mole?
Mole is Avogadro’s number (6.022 × 1023) of atoms, molecules, ions, or other species. Numerically, it is the atomic, molecular, or formula weight of a substance expressed in grams.
8. What is the formula for the number of moles of a substance?
Moles = grams/ formula weight (g/mol)
Millimoles= milligrams/ formula weight (mg/mmol)
9. How Do We Express Concentrations of Solutions?
Molarity
Normality
Formality
Molality
Density Calculations
10. What is Molarity?
The molarity of a solution is expressed as moles per liter or as millimoles per milliliter.
A one-molar solution is defined as one that contains one mole of substance in each liter of a solution. It is prepared by dissolving one mole of the substance in the solvent and diluting to a final volume of one liter in a volumetric flask.
11. What is Normality?
A one-normal solution contains one equivalent per liter.
An equivalent represents the mass of material providing Avogadro’s number of reacting units. A reacting unit is a proton or an electron. The number of equivalents is given by the number of moles multiplied by the number of reacting units per molecule or atom; the equivalent weight is the formula weight divided by the number of reacting units.
12. What is Formality?
Chemists sometimes use the term formality for solutions of ionic salts that do not
exist as molecules in the solid or in solution. The concentration is given as formal (F).
Operationally, formality is identical to molarity.
13. What is Molality?
A one-molal solution contains one mole per 1000 g of solvent. The molal concentration is convenient in physicochemical measurements of the colligative properties of substances, such as freezing point depression, vapor pressure lowering, and osmotic pressure because colligative properties depend solely on the number of solute particles present in solution per mole of solvent.
Molal concentrations are not temperature dependent as molar and normal concentrations are (since the solution volume in molar and normal concentrations is temperature dependent).
14. What is the difference between analyze and determine?
The terms analyze and determine have two different meanings. For example, a sample is analyzed for part or all of its constituents. The substances measured are called analytes. The process of measuring the analyte is called a determination.
15. What is Density Calculation?
Density is the weight per unit volume at the specified temperature, usually g/mL or g/cm3 at 20◦C. (One milliliter is the volume occupied by 1 cm3.)
16. What is Specific Gravity?
Specific gravity is defined as the ratio of the mass of a body (e.g., a solution), usually at 20◦C, to the mass of an equal volume of water at 4◦C (or sometimes 20◦C).
That is, specific gravity is the ratio of the densities of the two substances; it is a dimensionless quantity.
Since the density of water at 4◦C is 1.00000 g/mL, density and specific gravity are equal when referred to water at 4◦C. But normally specific gravity is referred to water at 20◦C; density is equal to specific gravity × 0.99821 (the density of water is 0.99821 g/mL at 20◦C).
Note: Density of solution at 20◦C = Specific gravity of solution × 0.99821 g/mL
17. What is the general formula for calculating percent on a weight/weight basis?

18. What is the formula for calculating parts per thousand (ppt), parts per million (ppm), or parts per billion (ppb)?
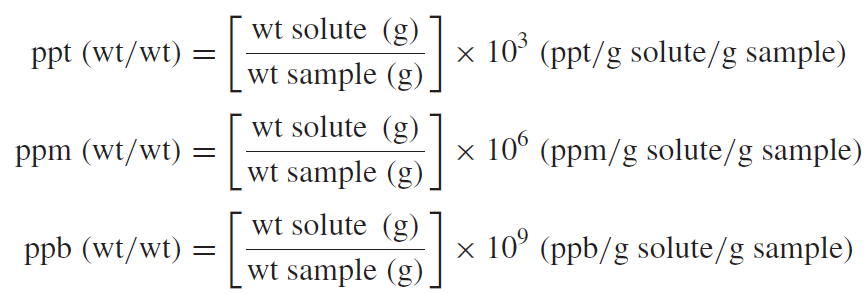
19. Explain the conversion from ppt (thousand), ppm, ppb, and ppt (trillion)?
1 ppt (thousand) = 1000 ppm =1, 000, 000 ppb; 1 ppm =1000 ppb = 1, 000, 000 ppt (trillion).
20. Explain the conversion from ppt (thousand), ppm, and ppb to weight/ weight?
ppt = mg/g = g/kg
ppm = μg/g = mg/kg
ppb = ng/g = μg/kg
21. Explain the Common Units for Expressing Trace Concentrations?
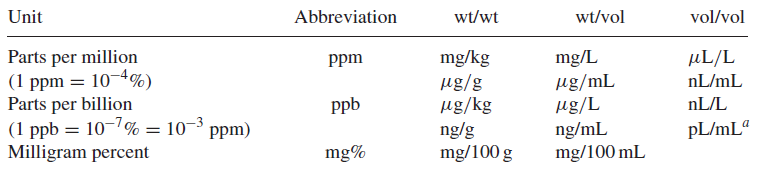

22. What is the formula for calculating percent, parts per thousand (ppt), parts per million (ppm), or parts per billion (ppb)?

23. Explain the conversion from ppt (thousand), ppm, and ppb to weight/ volume?
ppm = μg/mL = mg/L
ppb = ng/mL = μg/L
ppt = pg/mL = ng/L
24. What is the formula to calculate the milliequivalents of a substance from its weight in milligrams?

25. Explain Volumetric Calculations using Molarity?
a. Calculation for Gram
M (mol/L) × L = mol
g = mol × fw (g/mol)
g = M (mol/L) × L × fw (g/mol)
b. Calculation for milligram
M (mmol/mL) × mL = mmol
mg = mmol × fw (mg/mmol)
mg = M (mmol/mL) × mL × fw (mg/mmol)
26. How to calculate the percentage of an analyte that reacts on a 1:1 mole basis with the titrant?
%Analyte = fraction of analyte × 100% = mg of analyte/ mg of sample X 100%
= mmol analyte × fw of analyte(mg/mol) X × 100% / mg of sample
= M of titrant(mmol/mL) × mL of titrant × fw of analyte(mg/mmol) × 100% / mg of sample
27. What is Gravimetric analysis?
Gravimetric analysis usually involves the selective separation of the analyte by precipitation, followed by the very nonselective measurement of mass (of the precipitate).
28. What is volumetric, or titrimetric analysis?
In volumetric, or titrimetric, analysis, the analyte reacts with a measured volume of reagent of known concentration, in a process called titration.
29. What is Analytical Method Validation?
Analytical methods validation is a gauge taken to prove that the analytical methods employed for a specific test will produce results that consistently meet Predetermined specifications.
30. What is the impact of the unauthorized change in the analytical methods?
Changes in laboratory analytical methods may impact product quality and Regulatory commitments
31. What is the must before implementation of revised analytical method?
To implement the revised method, change control system requires and while proposing the change, revalidating the method prior to implementation of the change and release of product using the method is essential.
32. Which should be the minimum required document in the analyst training file?
Training files for laboratory analysts must contain the following a. SOP training documentation b. Analytical methods/ quality manuals training, and c. Analytical instrument training
33. Why Accuracy and precision is essential to be proved for the laboratory instruments?
The accuracy and precision of the analytical instrument play a vital role in the pharmaceutical industry to obtain valid data.
34. What are the essentials to maintain the consistent performance and operation of an instrument to perform within the operating and validated range?
The Qualification, calibration and preventive maintenance activities allows for laboratory instrument to continuously operate within operating parameters.
35. What is the process to track and maintain the instrument within the calibration state?
Instrument calibration schedule.
36. What types of traceability is required for calibration standards for each piece of laboratory instrument?
Calibration standards must be traceable to national or international standards that are acceptable to respective regulatory bodies who approved the GPS status of the company.
37. What minimum information should be available on reagent Labeled?
All reagents are Labeled with the date of receipt, the date the bottle is opened and the initials of the person opening.
38. From where the current lot of pharmacopoeia standards shall be verified?
Availability of correct lot number of all pharmacopoeia standards shall be ensured by periodic review of the corresponding Pharmacopoeia forum.
39. What should be ensured while collecting/ using the sample for analysis?
Product manufacturers need to ensure that any sample taken for analysis is true representative of the product, sample size and sample locations in case of powders, granules and liquids.
40. What should be the basis for deciding a sampling plan?
A sampling plan must be established with scientific justification and statistical criteria such as confidence levels, component variability, degree of precision desired, and the past history of the supplier.
41. How much sample should be collected for Raw Material and Finished Products?
The amount of samples taken must be sufficient for the quantity needed for analysis, retesting in case of OOS and Retention sample requirement as per the regulations.
42. What is the ideal quantity of retention samples?
The retention samples must be of twice the quantity necessary for all tests required to determine that the active ingredient meets its established specifications.
43. What is the first stage of the procedure in laboratory investigation to determine if the OOS result is observed?
The first stage of the procedure in laboratory investigation is to determine if the OOS result is laboratory error?
44. When laboratory investigation is inconclusive, what is the next stage of investigation?
If the result of the initial laboratory investigation is inconclusive, a full-scale laboratory investigation is required to be performed.
45. When OOS occurs, what to do with samples and reagents?
All the samples and reagents will be retained until the investigation has been completed.
46. What is calibration?
Calibration is a comparison between measurements of known magnitude and measurement made in as similar a way as possible with a second device.
47. What is the purpose of calibration?
The purpose of the GMP calibration requirements is to assure adequate and continuous performance of measurement instrument with respect to Accuracy and Precision and other applicable parameters.
48. What are the Colors of Different Wavelength Regions? Which Wavelength absorb which color?
| Wavelength Absorbed (nm) | Absorbed Color | Transmitted Color (Complement) |
| 380–450 | Violet | Yellow-green |
| 450–495 | Blue | Yellow |
| 495–570 | Green | Violet |
| 570–590 | Yellow | Blue |
| 590–620 | Orange | Green-blue |
| 620–750 | Red | Blue-green |
49. Explain wavelength, frequency, and wavenumber?
- The wave is described in terms of its wavelength, the distance of one complete cycle
- The wave is described in terms of the frequency, the number of cycles passing a fixed point per unit time.
- The reciprocal of the wavelength is called the wavenumber and is the number of waves in a unit length or distance per cycle.
50. What should be the choice of Solvents for Spectrometry?
The solvent used to prepare the sample must not absorb appreciably in the wavelength region where the measurement is being made.
51. What is Spectrometric Instrumentation?
A spectrometer or spectrophotometer is an instrument that will resolve polychromatic radiation into different wavelengths and measure the light intensity at one or more wavelengths.
Stability Studies
52. What is the purpose of stability studies?
The main purpose of performing stability studies is to collect information regarding the impact that environmental factors may have over the quality of drug substances and products and to determine the shelf life of products in specific container closure system at recommended storage condition.
53. What are the stability testing intervals for long term stability studies?
Stability study testing for long term stability studies shall be performed every three months
over the first year, every six months over the second year and annually thereafter.
54. What is the zero time point for stability sample analysis?
The time “zero” is defined as the first testing of the initial samples at the time of release.
55. When will the intermediate stage stability study be started?
The stability study procedure should address the requirement that in case when significant change observed at 40°C, 75%RH, then a study at 30°C/60% RH must be initiated and continued for 12 months.
56. What are the typical tests carried out during the stability studies of Small-volume parenterals?
Small-volume parenterals shall be tested for strength, appearance, colour, particulate matter, pH, sterility, and pyrogenicity.
57. What are the typical tests carried out during the stability studies of topical preparations in containers larger than 3.5 g?
All topical preparations in containers larger than 3.5 g will be sampled and tested at the surface, middle and bottom of the container to ensure homogeneity throughout the shelf life.
58. What are the typical tests carried out during the stability studies of Respiratory inhalers?
For Respiratory inhalers the specifications and testing requirements include delivered dose per actuation, number of doses, colour, clarity (solutions), particle size distribution (suspensions), loss of propellant, pressure, valve corrosion, and spray pattern.
59. Which ICH guideline is applicable for stability studies of drug substance and drug products?
ICH Q1A(R2), STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS
60. Why are stability studies carried out?
The purpose of stability studies is to provide scientific and documented evidence of how the quality of a drug product or drug substance changes with time under the influence of various environmental conditions such as humidity, temperature, and light. The study will help to establish a re-test period for the drug substance and shelf life for the drug product along with the
recommended storage conditions.
61. According to the ICH guideline, World can be divided into how many climatic zones?
World can be divided into four climatic zones, I-IV.
62. What is Stress Testing?
Stress testing is the testing required to be done to identify the likely degradation products or material, which can help to understand the degradation pathways and the inherent stability of the API/ Formulation. This will also help to validate the stability indicating capability of the analytical method used.
63. What parameters should be considered for stress testing?
During the stress testing, the effect of temperatures, humidity, hydrolysis condition, oxidation condition and impact of photoenergy should be considered.
i. Temperatures should be verified in 10°C increments – for example, 50°C, 60°C, 70°C etc. i.e. above that for accelerated testing.
ii. Humidity (e.g., 75% RH or greater) where appropriate, oxidation, and photolysis on the drug substance.
iii.Hydrolysis across a wide range of pH values when in solution or suspension.
iv. Light intensity
64. How many batches to be considered for stability testing of drug substance?
At least three batches of the drug substance.
65. How to select the test to be carried out for stability studies?
Tests to be considered for stability studies for the attributes that are susceptible to change during storage and are likely to influence quality, safety, and/ or efficacy. Stability specification should cover testing of physical, chemical, biological, and microbiological attributes.
66. What should be considered during method validation of the test method applicable for stability testing?
Analytical method validation for stability studies should be proved for its stability-indicating characteristics.
67. What should be the stability study testing frequency for new drug substances and drug products?
For long term conditions: Every 3 months over the first year, every 6 months over the second year, and annually thereafter.
For accelerated storage condition: Minimum of 3 time points, including the initial and final time points (Example – 0, 3, and 6 months).
Intermediate storage condition: This study shall be started if a result of significant change is observed at the accelerated storage condition. Minimum of 4 time points requires, including the initial and final time points (Example: 0, 6, 9, 12 months) and 12 month study duration is recommended.
68. What are the storage conditions for stability study for general case studies as per ICH Q1AR2?
Long term* – 25°C ± 2°C/60% RH ± 5% RH or 30°C ± 2°C/65% RH ± 5% RH (Minimum duration: 12 months)
Intermediate** – 30°C ± 2°C/65% RH ± 5% RH (Minimum duration: 6 months)
Accelerated – 40°C ± 2°C/75% RH ± 5% RH (Minimum duration: 6 months)
* It is up to the applicant to decide whether long term stability studies are performed at 25 2°C/60% RH 5% RH or 30°C 2°C/65% RH 5% RH.
**If 30°C 2°C/65% RH 5% RH is the long-term condition, there is no intermediate condition.
69. What are the storage conditions for stability study for the drug substances intended for storage at a refrigerator as per ICH Q1AR2?
Long term 5°C ± 3°C (Minimum duration: 12 months)
Accelerated 25°C ± 2°C/60% RH ± 5% RH (Minimum duration: 6 months)
70. What is the meaning of Significant change for drug substance?
The Significant change for a drug substance means failure to meet its specification.
71. How to deal with Significant change occurring at accelerated storage conditions during stability study for the drug substances intended for storage at a refrigerator?
When significant change observed between 3 and 6 months’ period at the accelerated condition, the re-test period should be proposed based on the long term data.
When, significant change observed within the 3 months’ study at the accelerated condition, it should be studied for potential effect of short term excursions outside the label storage condition, which could occur during storage, or shipping.
Short term study (less than 3 months with more frequent testing should be carried out, that would serve as a basis for justification for short term excursions outside the label storage condition.
72. What are the storage conditions for stability study for the drug substances intended for storage at a freezer as per ICH Q1AR2?
Long term – 20°C ± 5°C (Minimum duration: 12 months)
73. How to deal with Significant change occurring at accelerated storage conditions during stability study for the drug substances intended for storage at a freezer?
Since no accelerated stability study is guided in ICH guidelines for this storage condition, study on a single batch at an elevated temperature should be considered for data generation, for example, at 5°C ± 3°C or 25°C ± 2°C for a suitable time period. The purpose of the study is to address the effect of short term excursions outside the label storage condition that may occur during storage or shipping.
74. How is the stability study helpful in defining Labeling?
The statement regarding the storage condition of drug substance/ drug product should be based on the stability studies. Precautions should be added on the label based on the generated data, example, drug substances cannot tolerate freezing temperature.
75. What is the importance of Container Closure System in drug product stability study?
Stability testing for the drug product must be done with packaging material that are intended to market (including, as appropriate, any secondary packaging and container label). Container closure used for stability study should not be different from market pack, as container closure plays a vital role in protecting the quality of drug products.
76. What is a significant change for drug products?
Significant change for a drug product means:
1. A 5% change in assay from its initial results; or failure to meet the specification for potency when using immunological or biological procedures;
2. Any degradation product/ impurity or related substance exceeding its acceptance criterion;
3. Failure to meet the acceptance criteria for physical attributes, appearance, and functionality test (For example, phase separation, color, resuspendibility, hardness, caking, dose delivery per actuation); however, some changes in physical attributes may be expected under accelerated conditions; for example, melting of creams and softening of suppositories.
4. Failure to meet the acceptance criterion for pH; or
5. Failure to meet the acceptance criteria for dissolution for 12 dosage units.
77. What is the requirement of stability study for the drug products packaged in impermeable containers?
Stability studies for the drug product stored in impermeable containers can be done at any controlled or ambient humidity condition. (Note: Potential for solvent loss or Sensitivity to moisture is not an issue for drug products packed in impermeable containers as it provides a permanent barrier to passage of moisture or solvent.)
78. What is the additional requirement of stability study for the drug products packaged in semi-permeable containers?
When aqueous-based drug products are packed in semi-permeable containers, it should be evaluated for possibility of water loss in addition to other tests considered for stability studies.
79. What are the stability conditions considered for the drug products packed in semi-permeable containers?
Long term* 25°C ± 2°C/40% RH ± 5% RH or 30°C ± 2°C/35% RH ± 5% RH (Minimum Period: 12 months)
Intermediate** 30°C ± 2°C/65% RH ± 5% RH (Minimum Period: 6 months)
Accelerated 40°C ± 2°C/not more than (NMT) 25% RH (Minimum Period: 6 months)
*Applicants need to decide based on development data and region to where to be marketed for long term stability studies that are performed at 25 2°C/40% RH 5% RH or 30°C 2°C/35% RH 5% RH.
**If 30°C ± 2°C/35% RH ± 5% RH is the long-term condition, there is no intermediate condition
80. What is the significant change for loss in water for drug product packaged in a semi-permeable container?
A 5% loss in water from its initial value is considered a significant change for a product packaged in a semi-permeable container.
81. When 5% loss in water from its initial value may be considered justification for drug products packaged in a semi-permeable container?
For small containers (1 mL or less) or unit dose products, a water loss of 5% or more after an equivalent of 3 months’ storage at 40°C/NMT 25% RH may be appropriate, if justified.
82. Is intermediate stability testing needed when significant change for loss in water for drug products packaged in a semi-permeable container is observed?
A significant change in water loss alone at the accelerated storage condition does not necessitate testing at the intermediate storage condition.
83. What is Bracketing when referring to stability study?
Stability study design when only samples on the extremes of certain design factors, for example, strength, package size, are tested at all time points as in a full design called as Bracketing
84. Which ICH guidelines are applicable for Stability Testing?
ICH Q1A(R2): “Stability Testing of new drug substance and products”
ICH Q1B: “Photostability Testing of New Drug Substances and Products”
ICH Q1C: “Stability Testing of New Dosage Forms”
85. Why is photostability important?
To evaluate and demonstrate the impact of light exposure whether light exposure to the drug product or drug substance is resulting in unacceptable change or not.
86. Photostability testing is carried out on how many batches?
Photostability testing is carried out on a single batch.
87. As per ICH Q1B, what is the systematic approach to photostability testing?
A systematic approach to perform photostability testing is:
(i) Tests on the drug substance.
(ii) Tests on the exposed drug product outside of the immediate pack.
(iii) Tests on the drug product in the immediate pack.
(iv) Tests on the drug product in the marketing pack.
88. Which light sources are used for the photostability studies?
ICH Q1B, following are the two sources:
Option 1 – Light source that produces an output similar to the D65/ID65 emission standard. Example, artificial daylight fluorescent lamps combining visible and ultraviolet (UV) outputs, xenon, or metal halide lamp. For a light source emitting significant radiation below 320 nm, an appropriate filter(s) may be fitted to eliminate such radiation.
D65 is the internationally recognized standard for outdoor daylight as defined in ISO 10977 (1993).
ID65 is the equivalent indoor indirect daylight standard.
Option 2 – Same sample to be exposed to both the cool white fluorescent and near ultraviolet lamp.
1. A cool white fluorescent lamp produces an output specified in ISO 10977(1993) ;
2. A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm; a significant proportion of UV should be in both bands of 320 to 360 nm and 360 to 400 nm.”
Reference: STABILITY TESTING: PHOTOSTABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS Q1B
89. How to perform photostability testing of drug substances and drug product?
Photostability testing of drug substances and drug product consist of two parts -forced degradation (FD) testing and confirmatory testing.
The objective of FD testing is to evaluate the photosensitivity of the material for method development purposes and to understand the degradation pathway.
The objective of confirmatory studies are done to get the information necessary for handling, packaging, and labeling of drug substances.
90. How many batches shall be considered for the photostability of drug substance and drug product?
Generally one batch is tested during the development phase, and it is confirmed on a single batch. If the results of the confirmatory study are ambiguous, additional two batches should be considered for the study.
UV/ Visible spectrophotometry
91. What is the principle of UV/ Visible spectrophotometry?
The absorbance measured for an analyte can be linearly related to concentration (Lambert-Beer’s law).
92. What range of ultraviolet-visible (UV-vis) wavelengths are useful for UV-visible spectrophotometry?
Pharmaceutical applications of ultraviolet-visible (UV-vis) spectrophotometry concern light in the wavelength range 190–800 nm.
Ultraviolet (UV) range is from 190 to 400 nm
Visible region, recognized by the human eye, is from 400 to 800 nm.
93. What is Lambert-Beer’s Law?
Lambert law: Each layer of the medium through which the light is passing absorbs an equal fraction of light which is independent of the intensity of the incident light; thus, along the light path there is an exponential decay in the light intensity.
Beer’s law: Amount of light absorbed is proportional to the number of chromophores present in the medium that the light is passing through. It means the amount of absorbed light is proportional to the concentration of the absorbing species (chromophores).
94. What are the factors which influence the absorbance of molecules?
Absorbance of a molecule is dependent on the solvent, pH, molecular interactions, and temperature in addition to structure and wavelength.
These two laws are often combined into what is often referred to as Lambert-Beer’s,
Beer-Lambert’s or simply Beer’s law.
95. What are the factors which cause Deviation from Lambert-Beer’s Law and are the Sources of Error to induce deviation from the law?
Causes for deviation from Lambert-Beer’s law may be of chemical as well as instrumental origin
- At high drug or analyte concentrations (typically >0.01 M) causes deviations from linearity due to refractive index changes and because the close proximity of the absorbing molecules will affect their charge distribution and lead to alterations in their absorptivity.
- Particles present in the sample lead to light scattering.
- Polychromatic radiation
- Stray light. This light reaches the detector without having passed through the sample due to light scattering within the instrumentation or light entering from outside the instrument. The tray light will give negative deviations from Lambert-Beer’s law.
96. Why normally UV absorbance is measured at absorption maximum?
For quantitative analysis, a relatively narrow wavelength range where there is only a small change in absorptivity is selected which is normally found at the absorption maximum.
97. Explain the Instrumentation or basic components for UV/Visible spectrophotometry?
The basic components for UV/Visible spectrophotometry include a light source, a wavelength selector, a sample compartment (often a cuvette or flow cell) and a detector.
98. Explain which basic types of detectors are used in UV/Visible spectrophotometry?
Two basic types of detectors are used:
- Photomultiplier tubes
- Semiconductors
99. What are the types of UV/ Visible spectrophotometers?
- Single beam spectrophotometer
- Double beam spectrophotometer
- Array detector spectrophotometer
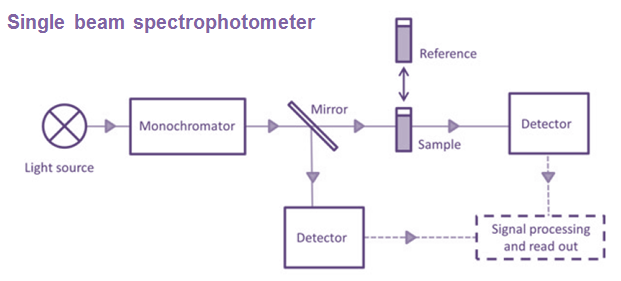


100. What are the applications of UV/Visible Spectrophotometry?
Applications of UV/Visible Spectrophotometry are:
- Qualitative Analysis
- Quantitative Analysis
- Quantitative Analysis – Use in physicochemical profiling of drug substances such as pKa determination and kinetic studies, Equilibrium Constants and Complexation – can be applied broadly to the characterization of equilibria and complexation phenomena, Kinetics and Reaction Monitoring, Dissolution Testing
Fluorescence Spectroscopy
101. What is Luminescence?
Luminescence is the spontaneous emission of radiation from a substance. Emission of radiation from species in electronically (or vibrationally) excited states not in thermal equilibrium with its environment.
102. What is Fluorescence Spectroscopy?
Fluorescence spectroscopy uses a light source to excite electrons in molecules and it emits light in visible range. The emitted light is detected using a detector for measurement and identification of the molecule. The concentration of solution is directly proportionate to the detector response.
103. What are the types of Luminescence?
The types of luminescence are classified according to the mode of excitation.
- Photoluminescence: When electromagnetic radiation (photons) energy gets absorbs, it emits Luminescence. Examples of Photoluminescence are:
- Phosphorescence: This is a delayed reaction. This happens when emission of photons is trapped in a ‘forbidden’ state. This action happens in milliseconds to hours.
- Fluorescence: This happens when rapid emission of photons as electrons jump from excited state to ground state.The actions happen in nanoseconds.
- Chemiluminescence: When chemical reaction emits Luminescence. Examples of Chemiluminescence are:
- Electrochemiluminescence: It occurs because of electrochemical reactions
- Bioluminescence: It occurs because of biochemical reactions in living organisms (For example – fireflies).
- Crystalloluminescence: Luminescence occurs while crystallization – This occurs when solid crystals precipitate from a solution, a molten material or deposited directly from a gas.
- Electroluminescence: Luminescence occurs while electric current passes from a substance. Type of Electroluminescence is Cathodiluminescence. This happens when electrons strike with a luminescent material.
- Mechanoluminescence: Luminescence occurs because of mechanical actions:
- Triboluminescence: Luminescence occurs when material is crushed, scratched, or rubbed. During this action, bonds in a material are broken when that
- Fractoluminescence: Luminescence occurs when bonds in crystals are broken by fractures.
- Piezoluminescence: Luminescence occurs when pressure generates on solids.
- Sonoluminescence: Luminescence occurs when imploding bubbles in a liquid whenever it is excited by sound
- Radioluminescence: Luminescence occurs by bombardment with ionizing radiation
- Thermoluminescence: Luminescence occurs on re-emission of previously absorbed energy while a substance is heated
Mid and Near Infrared Spectroscopy
104. What is the infrared (IR) spectroscopy spectral range?
Infrared (IR) spectroscopy has a spectral range between 12,500 and 20 cm-1. It is further subdivided in the:
- Far-IR (FIR: 400–20 cm-1)
- Mid-IR (MIR: 4000–400 cm-1)
- Functional group region (4000-1300 cm-1)
- Fingerprint region (1300-400 cm-1):
(Reference: https://byjus.com/chemistry/infrared-spectroscopy/)
- Near-IR (NIR: 12,500–4000 cm1)

Reference of diagram: https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Wade)/11%3A_Infrared_Spectroscopy_and_Mass_Spectrometry/11.05%3A_Infrared_Spectra_of_Some_Common_Functional_Groups
105. What is the principle of infrared (IR) spectroscopy?
When a molecule is exposed to electromagnetic radiation in the infrared (IR) region, it matches the frequency of its vibrational modes, absorbs the energy and jumps to a higher vibrational energy state. The difference in energy between the two vibrational states is equivalent to the energy associated with the wavelength of radiation that was absorbed. In this situation, the infrared region of the electromagnetic spectrum contains frequencies corresponding to the vibrational frequencies of organic bonds.
Absorbance in the MIR range originates from two types of fundamental vibrations, namely stretching and bending.
106. How is infrared (IR) spectroscopy provided information about the structure of a compound?
Answer: 1
The absorption of infrared radiation by a molecule causes changes in their vibrational and rotational energy levels, therefore, IR-spectroscopy is also known as vibrational-rotational spectroscopy. Similar to the UV-spectroscopy, very few peaks in their spectrum, IR spectroscopy provides a spectrum with a large number of absorption bands. Therefore, it provide plenty of information about the structure of a compound. Different bonds present in the spectra correspond to various functional groups and bonds present in the molecule.
Answer: 2
Frequency and intensity of the absorbed radiation depend on the strength of the bond, the atoms the molecule is composed of, the extent of the dipole moment change and the chemical environment. Hence, position, intensity and width of MIR absorption peaks provide information about the molecular structure of the sample including inter- and intramolecular interactions such as hydrogen bondings.
It deals with the absorption of radiation in the infrared region of the electromagnetic spectrum. IR spectrum gives sufficient information about the structure (identification of functional groups) of a compound and can also be used as an analytical tool to assess the purity of a compound.
107. What are the characteristics of mid and near infrared spectroscopy?
| Mid infrared spectroscopy | Near infrared spectroscopy | |
| Vibrations | Fundamentals | OvertonesCombinations |
| Wavenumber range | 4000–400 cm-1 | 12,500 –4000 cm-1 |
| Radiation (Light source) | Polychromonatic radiation (Globar tungsten) | Polychromonatic radiation (Globar tungsten) |
| Spectral principle | Absorption | Absorption |
| Absorption coefficient | High | Low |
| Absorbance peaks | Numerous and well resolved | Broad and overlapped |
| Selection rules | Change in dipole moment | Change in dipole momentAnharmonicity |
| Functionalities | Polar groups | X-H groups (i.e. CH/OH/NH groups) |
| Structural selectivity | High | Low |
| Quantitative measurements | Beer’s law | Beer’s law |
| Sample preparation | Dilution required (e.g. KBr)(except ATR-IR) | Not required |
| Sample size | Small volume (μl)Low thickness (μm) | LargeThickness up to cm |
| Monochromator Detection principles | FT-IR | GratingFT-IRAOTFDiode-array |
| Light-fiber optics | Chalcogenide or AgCl (<10m)Limited | Quartz (>100m) |
| Probes | ATR (attenuated total reflectance) | TransmissionTransflectionDiffuse reflectance |
108. What are the Infrared spectral regions within the electromagnetic spectrum?

109. What is the Functional group region (4000-1300 cm-1) and what is the significance?
In the functional group region (4000-1300 cm-1), most of the functional groups present in organic molecules exhibit absorption bands, therefore it is called a functional group region.
110. What is the Fingerprint region (1300-400 cm-1) and what is the significance?
The region from 1300-400 cm-1 has a complicated series of absorptions. These are mainly because of molecular vibrations, generally bending motions that are characteristic of the entire molecule or large fragments of the molecule.
Two different compounds will have different absorption patterns in this region except enantiomers. Therefore, the absorption patterns are unique in this region for any compound. For that reason, this region is called the fingerprint region.
111. Why Functional group region (4000-1300 cm-1) and Fingerprint region (1300-400 cm-1), both have an importance?
Two molecules having the same functional group could show the same spectra in the functional group region, however, their spectra differ in the fingerprint region. Therefore both the regions are very useful for confirming the identity of a chemical substance.
112. How are the sample preparations done for infrared spectroscopy?
Materials are available in the different forms for which the IR spectrum is recorded. The compounds are available in liquid, solid, gas and solution form.
Not all materials are transparent to IR rays, and few of them are opaque to IR radiation. Therefore, to obtain spectra, compounds must be dissolved or diluted in a transparent matrix.
Alkyl halides are compounds that are transparent to the IR region. The compound has the structure of C–X bond, where X is a halogen: bromine, chlorine, fluorene, or iodine. Generally the frequency of these bonds appear in the region 850-515 cm-1, which are out of the range of typical IR instrumentation.
Generally, the materials used for matrix (i.e. NaCl, KBr) are absorbs the moisture and water absorb IR rays near 3710 and 1630 cm-1 therefore the samples should be perfectly dried before utilization for the IR analysis,
(1) Solid samples: Solid samples are prepared using various methods, which are described as follows:
(i) Pressed disc: Solid sample is mixed with KBr and translucent pellet of this powder mixture is formed by pressing using mechanical pressure. KBr is transparent to IR radiation ranging from 4000-650 cm-1 hence, it gives adequate spectra without any interference. The demerit of using KBr is, it absorbs moisture quickly and that would interfere with the spectra.
(ii) Mull or paste: Sample is powdered and mixed with an oily mulling agent (typically
Nujol). The mixing is done with the help of mortar and pestle. A thin film of the mull is created, kept in between flat plates of NaCl and IR spectrum is recorded.
This method has demerit that nujol absorptions bands at 1380 cm-1, 1462 cm-1 and 2924-2860 cm-1, therefore no information about the observed compound can be obtained in this region.
(iii) Film: Dissolve the solid sample in non-hygroscopic solvent such as Carbon tetrachloride or Methylene chloride.
A drop of the above solution is deposited on the surface of the KBr or NaCl plate. The plate is dried by evaporation and film is formed. This KBr disc is used to obtain the IR spectrum.
(2) Liquid samples: Liquids are studied in solution or neat. A drop of neat liquid sample or a solution of the sample is kept between two plates of a NaCl or KBr to obtain a thin film. It is then analysed to obtain the spectrum.
The NaCl or KBr plates tend to break very easily and those are water soluble, hence, compounds to be analyzed must be free from water.
The spectra obtained using this method is also called as neat spectrum because no solvent is used while recording the spectrum.
(3) Gaseous samples: The gas is passed into a specially designed cell having a long path length (around 10 cm). The two side walls of the and the cell are made up of NaCl. The cell is placed inside the IR spectrophotometer to record the spectra.
113. What are the sampling techniques for measurements in the mid-IR?
There are three main sampling techniques for measurements in the mid-IR
(a) Transmission – Transmission measurements are useful with liquid and solid samples. Because of the high absorbance coefficient in the mid-IR region, solid samples are diluted with a non-absorbing substance such as KBr and pressed into a pellet.
(b) Attenuated total reflectance (ATR): In ATR technique, the sample is kept in direct contact with an internally reflecting material with a high index of refraction (e.g. ZnSe). The ray is transmitted through the ATR element is reflected at the crystal/ sample interface, thereby penetrating a few microns into the sample.
(c) Diffuse reflectance (DRIFT): In a DRIFT technique, the IR radiation reflected from rough sample surfaces is collected. The incident light is partly reflected by the sample surface (i.e. specular reflectance), partly scattered, and partly transmitted into the sample. The transmitted part may be absorbed or diffracted, resulting in diffusely scattered light.
Contrary to the specularly reflected part, which is usually eliminated by the DRIFT accessory, the diffuse reflected light collected over various angles thus contains absorptivity information from the sample. Due to the measurement principle, DRIFT spectroscopy can be used for noninvasive evaluation of solid samples including characterization of polymorphic forms.
114. What is Near infrared spectroscopy (NIR)?
Near infrared spectroscopy (NIR) is a technique of infrared spectroscopy where the spectrum of material is acquired by interaction between electromagnetic radiation and matter, within the wavelength range of 800 – 2500 nm or 12500 – 4000 cm-1. The IR radiation is absorbed those are related to different properties of the sample and provide quantitative and qualitative information.
The NIR range represents by weak overtones and combined bands emerged from the strong fundamental vibrations of C-H, O-H, C-O, C=O, C=O, N-H bonds and metal-OH groups in the mid-IR range.
115. What are the applications of NIR technology?
- Raw material identification
- Reaction Monitoring
- Crystallization
- Molecular Characterization of Solid Dispersions
- Challenges of Particle Size Determination
- Nondestructive Tablet Hardness Testing
- Nondestructive Prediction of Dissolution Performance
- Simultaneous Determination of Multiple CQAs
- Evaluation of Protein Secondary Structural Changes and Beyond
- Challenges of Antibody Formulations
- Noninvasive Analysis of Polymeric Protein Delivery Systems
- Use as Real-Time PAT Tool in Batch and Continuous Drug Product Manufacture – Blending, Wet Granulation, Fluid Bed Granulation, High shear wet granulation, Twin screw granulation, NIR in Roller Compaction, Hot-Melt Extrusion, Pelletization, Tableting and Capsule Filling, Coating, Freeze-Drying
- In Vivo Applications – Medical Monitoring, Tissue Analysis
116. What are the Infrared Sources?
The most common infrared sources are electrically heated rods of the following types :
(a) Sintered mixtures of the oxides of Zirconium (Zr), Yttrium (Y), Erbium (Er) etc., also known as ‘Nernst Glower’,
(b) Silicon Carbide ‘Globar’, and
(c) Various ceramic (clay) materials.
117. What are the Monochromators used in infrared spectroscopy?
(i) Metal Halide Prisms
(ii) NaCl Prism (2-15 μm)
(iii) Gratings
118. What are the Detectors used in infrared spectroscopy?
(a) Thermocouples (or Thermopiles): The underlying principle of a thermocouple is that if two dissimilar metal wires are joined head to tail, then a difference in temperature between head and tail causes a current to flow in the wires. In the infrared spectrophotometer this current shall be directly proportional to the intensity of radiation falling on the thermocouple. Hence, the thermocouples are invariably employed in the infrared region, and to help in the complete absorption of ‘available energy’ the ‘hot’ junction or receiver is normally blackened.
(b) Golay Detector : In this specific instance the absorption of infrared radiation affords expansion of an inert gas in a cell-chamber. One wall of the cell-chamber is provided with a flexible mirror and the resulting distortion alters the intensity of illumination falling on a photocell from a reflected beam of light. Thus, the current from the photocell is directly proportional to the incident radiation.
(c) Bolometers : These are based on the principle that make use of the increase in resistance of a metal with increase in temperature. For instance, when the two platinum foils are appropriately incorporated into a Wheatstone bridge, and radiation is allowed to fall on the foil, a change in the resistance is observed ultimately. This causes an out-of-balance current that is directly proportional to the incidental radiation. Just like the thermocouples, they are used in the infrared region.
Raman Spectroscopy
119. What is Raman spectroscopy?
Raman spectroscopy is a technique in which scattered light informs on the nature of the irradiated sample. Inelastic or Raman scattering happens when the changes in energy occurs during the collision between the molecule and monochromatic light, therefore, the frequency of the scattered light also changes. These changes provide information about the molecular identity and structure of the samples or material being analyzed.
120. What are the applications of Raman spectroscopy?
a. Active Pharmaceutical Ingredient (API) identification
b. Qualitative and Quantitative analysis of formulations
c. Detection of illicit substances
d. Monitoring of continuous manufacturing
121. What are the different types of Typical Raman Spectra, its advantages, disadvantages and area of applications?
| Category | Advantages | Disadvantages | Areas of application |
| FT-Raman | Low Fluorescence interference, quick test speed | Poor reproducibility caused by baseline drift | Quality control of traditional Chinesemedicine, identification of food fat, study of crystal morphology |
| Surface Enhanced Raman spectroscopy (SERS) | High sensitivity, trace detection | SERS effect is observed ona few substrates | Trace chemical detection of drugs such as methimazole and cannabinoids |
| Raman microspectroscopy (Rl) | Less sample required, high sensitivity, large information | Fluorescence interference | Physicochemical stability of dosage form, such as ibuprofen and crystal form studies |
| Resonance Raman spectroscopy (RRS) | Less sample required, high sensitivity: | Fluorescence interference | Drug interactions such as the doxorubicin and calf thymus DNA |
Source: https://www.iris-eng.com/
Thermal analysis techniques, Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA)
122. Explain Differential Scanning Calorimetry (DSC).
Differential Scanning Calorimetry (DSC) is a thermal analysis technique. It provides qualitative and quantitative information as a function of time and temperature regarding thermal transitions in materials that involve endothermic or exothermic processes, or changes in heat capacity.
123. What are the types of Differential Scanning Calorimetry (DSC)?
With respect to instrumentation, there are two main types of DSC instruments, power compensation and heat flux.
- Conventional DSC
- Power compensation DSC: This involves two separate furnaces for the reference and for the sample. The common principle of power compensation DSC is to heat both the reference and the sample simultaneously in such a way that the temperature of the two is kept identical, and the difference in power required to maintain the temperature is measured.
- Heat flux DSC: This instrument uses two crucibles for the sample and for the reference within one furnace. They are both heated from the same source and the temperature difference between the sample and the reference over the heating profile is measured.
- Modulated Temperature Differential Scanning Calorimetry (MTDSC)
Conventional DSC mentioned above is a powerful tool to measure a wide range of
thermal events such as melting accurately. However it often struggles to distinguish
overlapping thermal events such as overlapped glass transitions and endothermic
relaxation events, which can occur within the similar temperature range for many
amorphous drugs and polymers. MTDSC was designed to separate overlapping thermal events. Compared with conventional DSC, where a linear heating rate is applied, in MTDSC the sample follows a heating rate commonly with a sinusoidal modulated wave, and the uses of square and sawtooth modulated waves have also been reported.
- Hyper DSC
High speed or high performance conventional DSC, also known as hyper DSC, operates at extremely fast heating rates from 200 ° C/min up to 750 ° C/min. Conventional DSC using slow (linear) heating rates (typically heating rate below 100 °C/min) can result in good resolution but poor sensitivity particularly for phase transitions that strongly affected by kinetic factors, whilst fast heating rates can result in poor resolution but good sensitivity. Fast heating rates have the same total heat flow signal as in a DSC or MTDSC experiment. However as transitions occur over a shorter time period, the signal response to the thermal event appears larger. One issue that can occur with conventional DSC with slow heating is that the heating process may alter the sample, before the thermal transition of interest is reached. Using fast heating rates these effects can be eliminated or reduced, allowing for the characterisation of samples in their “as received” state. This technique is also of particular advantage for materials possessing properties that may change upon prolonged exposure to increased temperatures like amorphous products or formulations of biological molecules.
124. What is Thermogravimetric Analysis (TGA)?
Thermogravimetric Analysis (TGA) is one of the oldest thermal analytical procedures and has been used extensively in the study of material science.
The technique involves monitoring the weight change of the sample in a chosen atmosphere (usually nitrogen or air) as a function of temperature.
The measurement is operated by applying a temperature programme to a closed sample furnace containing an electronic microbalance (for holding the sample), which allows the sample to be simultaneously weighed and heated in a controlled manner, and the mass, time and temperature to be captured.
125. What are the applications of Thermogravimetric Analysis (TGA)?
- TGA is generally used for thermal stability and volatile components analysis.
- Evaluation of processing temperatures of thermally based manufacturing processes such as hot melt extrusion.
- It can determine the thermal stability of the drugs and polymers upon heating to assist with selecting the operation temperature in hot melt extrusion to avoid thermal degradation occurring in process.
- To measure the moisture or residue solvent contents in processed materials.
- TGA coupled with spectroscopic detection methods such as gas chromatography (GC and GC-MS) to allow the chemical identification of the volatile material liberated from the sample.
126. What is Scanning Probe Based Thermal Analysis?
The coupling of thermal analysis with atomic force microscopy (AFM) gives the new generation of thermal analysis the capability to allow thermal measurement to be performed at the selected point of interest. Such a technique is often known as localized thermal analysis (LTA).
127. What are the Physical and Chemical Phenomena Commonly Investigated Using Thermal Approaches?
a. Crystallization
b. Polymorphic Transformation
c. Glass Transition
d. Molecular Mobility
e. Structural Relaxation
f. Dehydration and Decomposition
128. What are the applications of Thermal Analysis for the Characterisation of Pharmaceutical Raw Materials?
a. Amorphous Drugs
b. Pharmaceutical Polymers and Lipidic Excipients
c. Polymer Blends
Data handling in analytical chemistry
129. What is meant by Accuracy?
Accuracy is the degree of agreement between the measured value and the true value.
130. What is meant by Precision?
Precision is defined as the degree of agreement between replicate measurements.

131. What is meant by the number of significant figures?
The number of significant figures can be defined as the number of digits necessary to express the results of a measurement consistent with the measured precision.
Weighing balance and weighing techniques
132. What are the types of weighing balances used in the quality control laboratory?
Single-pan mechanical balance
Semimicro balance
Microbalance
133. Explain classification and nomenclature of modern laboratory balances used in the quality control laboratory?
The classification and nomenclature of modern laboratory balances follows a decimal pattern based on the step size d of the associated digital display:
(i) Precision balances: d= 1, 0.1, 0.01, or 0.001 g
(ii) (Macro) analytical balances: d = 0.1 mg
(iii) Semimicro balances: d=0.01 mg
(iv) Microbalances: d = 0.00 1 mg
(v) Ultramicrobalances: d = 0.000 I mg
134. What are the typical sensitivity of Semimicro and Micro balances ?
The semimicro balance is sensitive to about 0.01 mg, and the microbalance is sensitive to about 0.001 mg (1 μg).
135. What is zero point drift in weighing?
The zero setting of a balance is not a constant that can be determined or set and forgotten. It will drift for a number of reasons, including temperature changes, humidity, and static electricity. The zero setting should therefore be checked at least once every half-hour during the period of using the balance.
136. What is the benefit of weighing in vacuum?
Weighing in vacuum gives more accuracy than weighing in air.
137. Explain the general rules of weighing?
i. Never handle objects to be weighed with the fingers. A piece of clean paper or tongs should be used.
ii. Weigh at room temperature, and thereby avoid air convection currents.
iii. Never place chemicals directly on the pan, instead weigh them in a vessel (i.e. weighing bottle, weighing dish, etc.) or on powder paper.
iv. Always brush spilled chemicals off immediately with a soft brush.
v. Always close the balance case door before making the weighing. Air currents will cause the balance to be unsteady.
138. How is the weighing of hygroscopic solid materials done?
Hygroscopic samples are weighed with the bottle kept tightly capped. Weighing by difference is required for hygroscopic samples.
139. How is the weighing of liquid done?
Weighing of liquids is generally done by direct weighing. The liquid is transferred to a weighed vessel (Example – a weighing bottle), which is capped to prevent evaporation during weighing, and is then weighed If a liquid sample is weighed by difference by pipetting out an aliquot from the weighing bottle, the inside of the pipet must be rinsed several times after transferring. Care should be taken not to lose any sample from the tip of the pipet during transfer.
140. What are the sources of error in weighing?
- Changes in ambient temperature or temperature of the object being weighed are probably the biggest sources of error, causing a drift in the zero or rest point due to convection-driven air currents. Hot or cold objects must be brought to ambient temperature before being weighed.
- Hygroscopic samples may pick up moisture, particularly in a high-humidity atmosphere.
- Exposure of the sample to air prior to and during weighing.
Volumetric glasswares
141. What are the examples of volumetric glassware?
VOLUMETRIC FLASKS
PIPETS (Transfer or volumetric pipets and Measuring pipets)

Transfer or volumetric pipets

Measuring pipets.
SYRINGE PIPETS

Hamilton microliter syringe.
BURETS

Titrations and standardization
142. What is titration?
In a titration, the test substance (analyte) reacts with an added reagent of known concentration, generally instantaneously. The reagent of known concentration is referred to as a standard solution.
It is typically delivered from a buret; the solution delivered by the buret is called the titrant. (In some instances, the reverse may also be carried out where a known volume of the standard solution is taken and it is titrated with the analyte of unknown concentration as the titrant.)
143. What is the equivalence point in titration?
The equivalence point is the theoretical end of the titration where the number of equivalents of the analyte exactly equals the number of equivalents of the titrant added. The end point is the observed end of the titration. The difference is the titration error.
144. What is the primary standard?
A standard solution is prepared by dissolving an accurately weighed quantity of a highly pure material called a primary standard and diluting to an accurately known volume in a volumetric flask.
145. What is the secondary standard?
A solution standardized by titrating a primary standard is itself a secondary standard. It will be less accurate than a primary standard solution due to the errors of titrations.
146. Explain the characteristics of the primary standard?
1. It should be 100.00% pure (0.01 to 0.02% impurity is tolerable).
2. It should be stable to drying temperatures, and it should be stable indefinitely at room temperature. The primary standard is always dried before weighing.
3. It should be readily and relatively inexpensively available.
4. If it is to be used in titration, it should possess the properties required for a titration. In particular, the equilibrium of the reaction should be far to the right so that a sharp end point will be obtained.
147. What is the classification of titration methods?
1. Acid–Base
2. Precipitation
3. Complexometric
4. Reduction–Oxidation
5. Non-aqueous titrations
6. Diazotization Titration
148. What is Acid–Base titration?
Both inorganic and organic, are either acids or bases and can be titrated using a standard solution of a strong base or a strong acid. Acid -base titrations are also called as neutralization or aqueous acid -base titrations.
The end points of these titrations are easy to detect, either by means of an indicator or by following the change in pH with a pH meter.
The acidity and basicity of many organic acids and bases can be enhanced by titrating in a nonaqueous solvent. The result is a sharper end point, and weaker acids and bases can be titrated in this manner.
149. Explain different theories of acid-base titrations.
Arrhenius Theory: The first theory was postulated by Arrhenius in 1884 as a part of general theory on electrolytic dissociation. As per this theory, acid is defined “as a substance which generates hydrogen ions when dissolved in water and further these hydrogen ions in association with solvent form hydronium ions’:

Bronsted Theory: Bronsted proposed the new theory in 1923; LOwry also proposed similar theory independently. He defined an acid as ‘a species that can donate protons’ and a base as
‘a species that can accept protons’.

In Bronsted theory, an acid donates proton. It is independent of solvent. This theory differs from Arrhenius theory in their concept of the base.
Lewis Theory: Lewis proposed a theory in 1923, called the Lewis theory. This theory describes an acid as a species which has the capability to accept an electron pair whereas a base is described as a species which has the capability to donate an electron pair. There is no change in the concept of acid-base as every proton acceptor is an electron-pair donor.
This theory is useful to describe the indicator colour change in non-protonic systems exhibiting acid-base reaction.
Law of Mass Action:
The law of mass action was proposed by Goldberg and Wage in 1867. The basics of this law are about the mass of the substances that react in a reaction. The law states that “The rate of a chemical reaction is proportional to the active masses of the reacting substances”.
In dilute solutions where conditions approach the ideal state, ‘active mass’ may be represented by the concentration of the reacting substances, i.e. gm-molecules or gm-ions per litre. The constant of proportionality is known as ‘velocity constant’.
150. What is Strong Acid or Base?
A strong acid is completely dissociated into its component ions in dilute aqueous solution.
Examples:
Strong acids: HCl, HClO4HNO3
Strong bases: NaOH, KOH
151. What is the Buffer solution?
Buffer solution is a solution of substance or a mixture of substances which helps in maintaining and establishing specific pH.
152. What are Neutralization Indicators?
These are the substances used in acid -base titrations that are helpful in detection of end point at the end of reaction. They generally exhibit different colours at the end point at various values of pH. These Indicators exhibit some colour in acidic pH whereas when the pH changes, it produces different colours. So after the neutralization point, they produce colour change as per the pH of the titrant or titrate, and thus denotes the end point. They are weak acids or weak bases, which have different colours in their conjugate base and acid forms. Most indicators are used in dilute solution form.
153. Explain the theories of Acid-Base Indicators?
(A) Ostwald Theory: W. Ostwald postulated the first theory to expla in the behavior of indicators. As per this theory, the undissociated indicator acid or a base has a colour different from its ion.
| Titration between | pH at end point | Commonly used indicators |
| Weak acid and Strong base | Alkaline range | Thymol blue,Phenolphthalein,Thymolphthalein |
| Weak base and Strong acid | Acidic range | Methyl orange,Methyl red,Bromocresol green |
(B) Resonance Theory: This theory explains that the acid-base indicators in use are usually organic compounds. However, they produce different colours in acid and base medium.
| Indicators | pH Range | Acid | Alkaline |
| Methyl orange | 03.1-04.4 | Red | Orange |
| Thymol blue | 01.2-02.8 | Red | Yellow |
| Bromophenol blue | 03.0-04.6 | Yellow | Blue |
| Methyl red | 04.2-06.3 | Red | Yellow |
| Phenolphthalein | 08.3-11.0 | Colourless | Red |
| Phenol red | 06.8-08.4 | Yellow | Red |
| Bromocresol green | 03.8-05.4 | Yellow | Blue |
154. What is Precipitation titration?
In precipitation titration, the titrant generates an insoluble product with the analyte. Indicators can be used to detect the end point, or the potential of the solution can be monitored electrically.
155. What are the different ways through which the separation can be achieved?
There are different ways through which the separation can be achieved which are given below.
1. Precipitation method.
2. Volatilization or evolution method.
3. Electro-analytical method.
4. Miscellaneous physical methods.
156. Explain the principle and steps involved in gravimetry?
Principle:
As already described, the principle involved in gravimetry is the quantitative estimation of component on the basis of measurement of mass. More precisely, the mass of an ion in a pure compound can be determined using gravimetry which is then applied to determine the mass percent of the same ion in a known quantity of a sample or impure compound.
The steps involved in the practice of gravimetry are shown below:
1. Preparation of a solution containing a known weight of the sample.
2. Separation of the desired constituent.
3. Weighing the isolated constituent.
4. Computation of the amount of the particular constituent in the sample from the observed weight of the isolated substance.
Steps Involved in Gravimetry:
The steps involved in practice of Gravimetric analysis are explained below:
1. Sample preparation.
2. Preparation of solution or dissolution.
3. Precipitation.
4. Testing the completeness of precipitation.
5. Digestion or Ageing of precipitate.
6. Filtration.
7. Washing of precipitate.
8. Drying or ignition of precipitate.
9. Weighing.
10. Calculations.
157. What is Complexometric titration?
In complexometric titrations, the titrant is a reagent that generates a water-soluble complex with the analyte (i.e. metal ion). Chelating agent is generally used as titrant.
Ethylenediamine Tetraacetic Acid (EDTA) is one of the most useful chelating agents used for titration. EDTA reacts with a large number of metal ions, and the reaction is controlled by adjustment of pH. Indicators used to detect the end point are forming a highly colored complex with the metal ion.
158. Classify the Complexometric titration?
- Complexometric titrations are classified into the following categories:
- Direct titrations
- Back titrations
- Replacement titrations
- Alkalimetric titration of metals
- Indirect titrations
159. What is Non-aqueous titration?
Non-aqueous titration involves the reaction between acid and base in presence of non-aqueous i.e. organic solvents.
160. Categorize Non-aqueous titration?
The non-aqueous titrations can be categorized mainly in two classes; e.g.
(a) Acidimetry: Some substances behave as base under the condition of titrations, thus determination of basic substances is categorized as acidimetry.
(b) Alkalimetry: Some substances behave as acid under the conditions of titrations, thus determination of acidic substances is categorized as alkalimetry.
161. What is Reduction-Oxidation or redox titration?
In Reduction-Oxidation or redox titration, an oxidizing agent with a reducing agent, or vice versa are used.
An oxidizing agent gains electrons and a reducing agent loses electrons in a reaction between them. Suitable indicators are available to detect the end point or various electrometric means are available to detect the end point.
162. What is Diazotization Titration?
This titration involves the conversion of the primary aromatic amine to a diazonium compound by the reaction with sodium nitrite. In this method, the primary aromatic amine is reacted with the sodium nitrite in acidic medium to form a diazonium salt.
Gas Chromatography (GC)
163. What is Gas Chromatography (GC)?
Gas chromatography is the method of compound separation from a mixture by injecting a liquid or gaseous sample into a mobile phase. The mobile phase is also called the carrier gas. The gas passes through a stationary phase.
In gas chromatography, the sample is converted to the vapor state (if it is not already a gas) by injection into a heated port, and the eluent is a gas (the carrier gas).
The stationary phase is generally a nonvolatile liquid or a liquid-like phase supported on or bonded to a capillary wall or inert solid particles such as diatomaceous earth.
164. What is the characteristic of the mobile phase in Gas Chromatography?
In the gas chromatography, the mobile phase is generally an inert (or unreactive gas). For example, argon, helium, nitrogen or hydrogen.
165. Explain stationary phase of Gas Chromatography?
The stationary phase of Gas chromatography is a microscopic layer of viscous liquid that is layered on the surface of solid particles. The solid particles are further adhered on an inert solid support inside a piece of glass or metal tubing. This is also called the Gas Chromatography column. In some cases, the surface of the solid particles in the CG column act as the stationary phase.
166. Explain the types of Gas Chromatography columns?
There are two types of columns used in Gas Chromatography (GC):
i. Packed columns
ii. Capillary columns
Packed columns came first and were used for many years.
Capillary columns are more commonly used today, but packed columns are still used for applications that do not require high resolution or when increased capacity is needed.
167. Explain the types of Gas Chromatography?
There are two types of GC.
i. Gas–solid (adsorption) chromatography
ii. Gas–liquid (partition) chromatography
168. What Compounds Can Be Determined by GC?
The compound to be determined on GC must be volatile and stable at operational temperatures, typically from 50 to 300◦C.
GC is useful for:
● All gasses
● Most nonionized organic molecules, solid or liquid, containing up to about 25 carbons
● Many organometallic compounds (volatile derivatives of metal ions may be prepared)
If compounds are not volatile or stable, often they can be derivatized to make them amenable to analysis by GC. GC cannot be used for macromolecules nor salts, but these can be determined by HPLC and ion chromatography.
169. What are the types of Gas Chromatography Detectors and its applications?
| Sr. No. | Detector | Application |
| 1. | Thermal conductivity | General, responds to all substances |
| 2. | Catalytic combustion | Very similar to the FID |
| 3. | Flame ionization | All organic substances; some oxygenated products respondpoorly. Good for hydrocarbons |
| 4. | Flame photometric | Sulfur compounds (393 nm), phosphorus compounds (526 nm) |
| 5. | Flame thermionic | All nitrogen- and phosphorus containing substances |
| 6. | Rubidium silicate bead | Specific for nitrogen- and phosphorus-containing substances |
| 7. | Argon ionization (β-ray) | All organic substances; with ultrapure He carrier gas, also for inorganic and permanent gasses |
| 8. | Electron capture | All substances that have affinity to capture electrons; no response for aliphatic and naphthenic hydrocarbons |
| 9. | Vacuum UVabsorption | Nearly all substances but inert gasses and nitrogen |
| 10. | Mass spectrometry | Nearly all substances. Depends on ionization method |
Techniques of pharmaceutical analysis
170. What are the various techniques of pharmaceutical analysis?
The techniques of pharmaceutical analysis can be divided into two major categories.
i. Qualitative analysis
ii. Quantitative analysis
171. What is Qualitative Analysis?
Qualitative Analysis involves various test procedures that are designed for the identification of compounds in the sample. These test results confirm the presence or absence of a compound in the sample to be analyzed.
172. What is Quantitative Analysis?
Quantitative analysis involves the quantitative determination of compounds in the sample. Quantitative analytical techniques are further classified as follows:
i. Chemical Methods: (a) Volumetric, (b) Gravimetric, (c) Gasometric
ii. Physico-chemical Methods or Instrumental Methods
iii. Microbiological Methods
iv. Biological Methods
173. What is the Volumetric Method of Analysis?
In volumetric methods, measurement of volume of solution is taken as a parameter for assay.
The volume of known strength of a solution that is required to react completely with the substance to be analyzed is measured. The quantity of analyte is determined from the volume of solution by calculation. The solution or reagent is called as titrant and the analyte to be analyzed is termed as titrate.
174. What are the types of Volumetric Method of Analysis?
Volumetric methods are classified into different types depending upon the type of reactions involved in the reaction which are as follows:
i. Neutralization titrations
ii. Non-aqueous titrations
iii. Precipitation titrations
iv. Oxid at ion-reduction titrations
v. Complexometric titrations.
175. What is the Gravimetric Method of Analysis?
In Gravimetric analysis, quantitation is done on the basis of weight of compound. This process involves isolation and weighing of the compound of known composition, i,e. purest form.
The analysis is carried out by various processes such as precipitation, volatilization, electro-analytical etc.
176. What is the Gasometrical Method of Analysis?
In Gasometrical method, the measurement of the volume of gases forms the basics of analysis.
When a chemical reaction is carried out under the specific process, the volume of gas evolved or absorbed in the reaction is measured. The measured volume is corrected to standard conditions of temperature and pressure. Gas burettes or nitro-meters are used for the measurement of volume of gas. Example, for gases that are measured by gasometrical methods are carbon dioxide, cyclopropane, oxygen, nitrous oxide, octal nitrate, nitrogen, amyl nitrate, ethylene and helium.
177. What is the Instrumental Method of Analysis?
Instrumental Method of Analysis involves the usage of instruments to measure the physical or physicochemical property of the compound to be analysed thus lead to quantitation of the compound. Following are the examples.
| Physical Properties | Instrumental Methods |
| Electrical potential | Potentiometer |
| Electrical conductance | Conductometry |
| Electrical current | Polarography and voltammetry |
| Absorption of radiation | Spectrophotometry, Colorimetry,Atomic absorption spectroscopy |
| Emission of radiation | Emission spectroscopy,Flame photometry,Fluorimetry |
| Scattering of radiation | Turbidimetric and Nephelometry |
| Refraction of radiation | Refractometry |
| Rotation of plane polarized light | PolarimetryOptical rotatory dispersion |
| Thermal properties | Thermal method of analysis(DSC, DTA, TGA) |
| Mass to charge ratio | Mass spectrometry |
| Separation | Chromatography. |
178. What is the Microbiological Method of Analysis (with example of antibiotic)?
Microbiological Method of Analysis for antibiotics involves the determination of inhibition of growth of bacteria by the substances to be analysed in comparison with the standard compound. On the basis of the result, the therapeutic efficacy of the antibiotics are decided.
The methods which are generally used include the cylinder plate (or cup plate) method and the Turbidimetric (or tube assay) method.
179. What is the Biological Method of Analysis?
Biological assays are carried out to observe the biological effect of the drug on some type of living matter. They are also called as bioassays.
Preparation and standardization of various molar and normal solutions
180. Preparation of N/10 Oxalic Acid.
Oxalic acid (COOH),.2H,O is to be dissolved in one litre of distilled water to get N/10 oxalic acid solution.
Weigh accurately 6.3 g of oxalic acid [(COOH)2 2H2O] and transfer it into a volumetric flask (1 litre), half-filled with distilled water. Shake well and make the volume up to the mark.
Note: If anhydrous oxalic acid (COOH)2 is available, then dissolve 4.5 g of the acid in one litre of distilled water to get 0.1 N oxalic acid solution.
181. Preparation of N/10 NaOH Solution.
Molecular weight of NaOH = 40
Acidity (number of replaceable OH group) = 1
Equivalent weight of NaOH = 40
Therefore, 4 g of NaOH dissolved in one litre of solution will give N/10 NaOH solution.
Procedure:
Weigh accurately 4 g of NaOH in a beaker and dissolve it in distilled water. Weighing should be performed quickly as it is hygroscopic. Transfer the contents and the washings to a 1 litre volumetric flask. Cool and then make the volume up to the mark. Shake well.
Standardization:
The N/10 NaOH prepared as per the above mentioned procedure is standardized by titrating against N/10 oxalic acid using phenolphthalein as an indicator. 10 ml of N/10 oxalic acid is taken in a conical flask to which 2-3 drops of phenolphthalein is added and mixed well. This solution is titrated slowly with constant stirring against N/10 NaOH taken in a burette. Titration is continued till the appearance of permanent pale pink colour as the end point. The volume of approximate N/ 10 NaOH solution at the end point is taken for calculation of normality of NaOH using the following formula.
N1V1 = N2V2
(Base) (Acid)
N1 – Normality of NaOH solution. (?)
V1 – Volume of NaOH solution used. (ml)
N2 – Normality of standard oxalic acid solution. (0.1 N)
V2 – Volume of standard oxalic acid solution. (10 ml)
182. Potassium Permanganate.
Preparation of N/10 KMnO4 Solution:
Molecular weight of KMnO4 = 158 g/mol
Equivalent weight of KMn04 is reaction specific. In acidic medium KMn04 is used as an oxidiser. So there will be 5 electrons gained by the Mn atom. Hence, the equivalent weight of KMn04 = Molecular weight/Number of electrons gained in redox reaction = 158/5 = 31.6. So 3.16 or 3.2 g of KMnO4 is weighed accurately and dissolved in 1 litre of distilled water to get N/10 KMnO4 solution.
In alkaline or neutral medium, reaction of KMnO4 is different and Mn gains 3 electrons in redox reaction. So, for alkaline medium redox titrations, equivalent wt of KMnO4 will be 158/3 = 52.6.
So for 0.1 N KMn04 solution in alkaline medium redox titration, 5.26 g of KMn04 is weighed and dissolved in 1 L distilled water.
Procedure:
In general, 3.2 g of KMn04 is accurately weighed and dissolved in one litre of distilled water. The solution is boiled for 10-15 minutes and then allowed to stand for few days and filtered through glass wool.
Standardization:
10 ml of N/10 oxalic acid is taken in a conical flask. Add 5 ml dilute sulphuric acid, warm it to 60 -70 C and titrate against KMnO4 from the burette till a light pinkish colour appears.
Repeat the titration until concomitant results are obtained. The strength of KMnO4 is calculated using the formula N1V1 = N2V2
Example:
Suppose 10 ml 0.1 N oxalic acid = 8.5 ml of KMnO4
N1V1 = N2V2
0.1 N x 10 = N2 x 8.5
N2 = (10 x 0.1)/8.5=0.1176
To prepare 1000 ml 0.1 N KMnO4, the volume of KMnO4 taken is, (1000 x 8.5 x 0.1)/10 x 0.1 = 850
Now, take 850 ml of prepared KMn04 solution and make it 1000 ml by adding distilled water.
Note: Ordinary or even pure distilled water contains traces of organic matter which reduces the KMnO4 solutions. That is why the solution is boiled and kept for some time before standardization. In the absence of sufficient amount of dilute H2SO4 or due to the rapid addition of KMnO4 in titration flask, brown turbidity (manganous oxide) may appears.
183. Sulphuric Acid.
Preparation of N/10 H2SO4
Equivalent weight of H2SO4 = 49 g
Specific gravity = 1.84 g/ml
So, volume of 49 g H2SO4 = 26.6 ml
Concentrated H2SO4 (reagent grade) is about 97% pure.
Therefore, the actual amount of concentrated H2SO4 required for 1.0 litre of N/10 H2SO4 solution = (100/97) x 26.6 = 27.42 ml.
Thus, for 1.0 litre of N/10 H2SO4 solution, 2.74 ml of concentrated H2SO4 is required.
Procedure:
Take 2.74 ml sulphuric acid in a beaker filled with a small amount of distilled water. Transfer the contents of the beaker to a volumetric flask of 1 litre capacity and make volume up to the mark with distilled water. Shake well.
Standardization:
N/10 H2SO4 is titrated with 10 ml of 0.1 N Na2CO3 using mixed methyl orange as an indicator. Repeat the titration until at least three concordant readings are obtained.
Suppose, 10 ml of 0.1 N Na2CO3 = 9.5 ml of H2SO4
V1N1 = V2N2
10 x 0.1 = 9.5 x N2
N2 = 0.10526
To prepare 1 litre N/10 H2SO4, the volume of 0.10526 N acid required is (1000 x 0.1)/ 0.10526 = 950 ml.
Take 950 ml of 0.10526 N acid and dilute it to one litre. Check it again with N/10 Na2CO2
for three times. It must neutralize an equal volume of N/10 Na2CO3 solution. Label it as 0.1 N H2SO4
184. Hydrochloric Acid.
Preparation of N/10 HCl:
Molar mass for HCI is 36.4611 g/mol. Since HCI has only one hydrogen ion, the equivalent mass will be 36.4611. Specific gravity for 1 litre volume of HCI is 1.189.
For 1 litre volume, Grams of compound needed = (0.1 N)(36.4611)(1 Litre) = 3.6461
Volume of concentrated acid (37.5%) needed =3.6461/(0.375 x 1.189) = 8.1774 ml
So, 8.1774 ml of 37.5% concentrated HCI is dissolved in 1 litre of water to prepare 0.1 N HCI.
Procedure:
Transfer exactly 20 ml of the approximately 0.1 M HCl solution into a 250 ml conical flask.
Add 3 drops of phenolphthalein as an indicator. Titrate against standard N/10 NaOH solution until a permanent pale pink colour appears. Using the volume of NaOH, the strength of HCI is calculated.
Alternatively, HCI can be standardized by titrating with standard N/10 Na2CO3 using methyl orange as indicator. Colour change: yellow to reddish orange
Reaction equation: Na2CO3(aq) + 2 HCI(aq) —–> 2 NaCI(aq) + H2O (l) + CO2(g)
Standardization:
HCI is standardized against standard N/10 NaOH which is already standardized against
N/10 oxalic acid using Phenolphthalein indicator.
HCI + NaOH —-> NaCI + H2O
185. Preparation of 0.1 N Sodium Thiosulphate Solution (Na2S2O3.5H2O).
Dissolve approximately 24.8 gm of sodium thiosulphate crystals in previously boiled and cooled distilled water and make the volume to 1000 ml.
Store the solution in a cool place in a dark colored bottle.
After storing the solution for about two weeks, filter if necessary and standardize as follows:
Standardization:
Weigh accurately about 5.0 gm of finely ground potassium dichromate which has been previously dried to a constant weight at 105 ± 2°C into a clean 1.0 litre volumetric flask. Add sufficient distilled water to dissolve the content of volumetric flask and make up to the mark with distilled water; shake thoroughly and keep the solution in a dark place. Pipette 25.0 ml of this solution into a clean glass stoppered 250 ml conical flask. Add 5.0 ml of concentrated hydrochloric acid and 15.0 ml of 10% potassium iodide solution. Allow to stand in dark for 5 minutes and titrate the mixture with the solution of sodium thiosulphate using starch solution as an indicator towards the end point. The end point is taken when blue color changes to green. Calculate the normality (N) of the sodium thiosulphate using formula.
186. Preparation of 0.1 N Ceric Ammonium Sulphate.
Dissolve 66 gm of eerie ammonium sulphate with gentle heat in a mixture of 30 ml of sulphuric acid and 500 ml of water. Cool the mixture, filter and dilute to 1000 ml with water.
Standardization of 0.1 N Ceric Ammonium Sulphate:
Arsenic trioxide is allowed to dry for an hour. From this, weigh about 0.2 gm of Arsenic trioxide accurately and transfer into a sao ml conical flask. Wash the inner walls of the conical flask with 100 ml of water and mix thoroughly. To this, add 300 ml of dilute sulphuric acid, 0.15 ml of osmic acid and 0.1 ml of ferrous sulfate indicator. Titrate this solution with eerie ammonium sulphate sa luting which was taken in the burette. Continue the titration till the pink colour of the solution changes to pale blue or yellowish green colour. Each ml of 0.1 N ceric ammonium sulphate ~ 0.6326 gm of ceric ammonium sulphate – 4.946 grams of arsenic trioxide.
Errors in pharmaceutical analysis
187. What are the categories of errors that could occur during the analysis?
Two major categories of errors are known as (i) absolute error and (ii) relative errors.
(i) absolute error: The difference between the experimental mean and a true value is known as ‘absolute error’.
(ii) relative errors: The relative error is the value found by dividing the absolute error by the true value.
Relative error = (Measured mean value – True value)/ True value
188. What are the types of error based on its source?
Depending on the nature of errors which affects the accuracy or precision of a measured quantity, it is classified as follows:
i. Determinate Errors: These are ascertainable errors that can be either avoided or corrected. The error may be constant as in the case of weighing with uncalibrated weights or in measuring a volume using burette or pipette. Such measurable determinate errors are categorized as systematic errors.
ii. Indeterminate Errors: Indeterminate errors are often called accidental or random errors. They are revealed by small differences in a series of measurements made by the same analyst under identical conditions.
Determinate Errors are as follows:
Instrumental errors – Faulty equipment or low quality equipment
Personal errors – These errors occur by persons who are handling the method of analysis. The error may be resulted due to carelessness or ignorance and even by unskilled persons. This error is also called operative error.
Chemical errors – These errors are resulted by using chemicals and reagents with impurities or contaminants which may interfere with the reactions, thus affects the results.
Errors in the methodology – This is a most serious error in analysis as the error arises due to faulty method, e.g. co-precipitation of impurities, slight solubility of precipitate, incomplete reactions etc. Errors of this category are usually detectable and can be eliminated to a large extent.
189. How to minimize errors?
Methods to minimize the errors are discussed as follows:
1. Instrumental Errors: Instrumental Errors can be minimized by checking thoroughly the equipment used for the analysis before starting any analysis. Proper calibration should be performed to ensure the performance of equipment. Faulty equipment should be corrected by experts and rechecked for accuracy of results. If the performance is not satisfied then replacement should be done.
2. Personal Errors: Trained persons should perform analysis. Regular reporting and monitoring or analysis can be done.
3. Chemical Errors: Standard chemicals from authentic sources without impurities must be used for the analysis. The quality of chemicals and reagents can be checked periodically as per the standard guidelines.
4. Errors in Methodology: These errors can be avoided by following the standard methods with proper references. Continuous monitoring of reactions by skilled persons can be employed to minimize these errors
5. Indeterminate Errors: Since indeterminate errors are not predictable, the entire procedure of analysis should be carried out in a well -planned manner considering all factors which affect the accuracy and precision of the results.
Significant figures
190. What is a significant figure?
The number of significant figures can be defined as, “the number of digits necessary to express the results of a measurement consistent with the measured precision”.
Or
“the number of digits necessary to express the results of a measurement that is inline with the specification”.
Pharmacopoeia
191. What is Pharmacopoeia?
The word Pharmacopoeia derives from the ancient Greek word pharmakopoiia. Pharmako denotes “drug “; whereas the word (poi-) denotes “make”, thus the term collectively denotes “drug-mak-ing” or “to make a drug”.
A pharmacopoeia, pharmacopeia, or pharmacopoea, is a legally binding collection of standards and quality specifications for medicines used in a country or region. It is prepared by a national or regional authority in that country or region.
192. What is the content available in the Pharmacopoeia?
The pharmacopoeia comprises the recommended procedures for analysis and specifications for the determination of pharmaceutical substances, excipients and dosage forms and thus maintains the quality of medicines.
Most of the pharmacopoeias consist of a general part which includes tests, methods and general requirements for pharmaceutical substances and a specific part which is designed in the form of monographs for pharmaceutical substances.
193. What should be the choice of method between pharmacopoeia or in-house?
Pharmacopoeial method should be method of choice instead of in-house method of analysis.In case pharmacopoeial method does not work it should be justified and scientifically establish the rationale that in-house method is superior then the pharmacopoeial method for the said formulation
194. What should be the frequency of pharmacopoeial method revision?
All pharmacopoeias are needed to be updated constantly due to the continuing scientific progress. The method adopted by an organization also needs to be updated inline with pharmacopoeia as per the given timeline by pharmacoloeia.
195. Who publishes pharmacopoeia?
National level pharmacopoeia refers to pharmacopoeias which are published by the national pharmacopoeial commission of individual countries.
196. What are the pharmacopoeial editions of Indian Pharmacopoeia (IP)?
| Edition | Year | Volumes | Addendum/ Supplement |
| 1st Edition | 1955 | – | Supplement 1960 |
| 2nd Edition | 1966 | – | Supplement 1975 |
| 3rd Edition | 1985 | 2 | Addendum 1989 Addendum 1991 |
| 4th Edition | 1996 | 2 | Addendum 2000 Vet Supplement 2000 Addendum 2002 Addendum 2005 |
| 5th Edition | 2007 | 3 | Addendum 2008 |
| 6th Edition | 2010 | 3 | Addendum 2012 |
| 7th Edition | 2014 | 4 | Addendum 2015 Addendum 2016 |
| 8th Edition | 2018 | 4 | Addendum 2019 Addendum 2021 |
| 9th Edition Effective date is 1st December 2022 (Tentative) | 2022 | – |
Impurities in the pharmaceuticals
197. What are the Sources of Impurities in the pharmaceuticals?
- Raw Material Employed in Manufacture
- Method or the Process used in Manufacture
- Chemical Processes and Plant Materials employed the Process
- Storage Condition
- Decomposition
198. What are the effects of Impurities in pharmaceuticals?
Effects of Impurities in pharmaceuticals are:
1. Impurities which have a toxic effect can be injurious when present above certain limits.
2. Impurities, even when present in traces, may show a cumulative toxic effect after a certain period.
3. Impurities are sometimes harmless, but are present in such a large proportion that the active strength of the substance is lowered. The therapeutic effect of drugs is decreased.
4. Impurities may bring about a change in the physical and chemical properties of the
substance, thus making it medically unfit.
5. Impurities may cause technical difficulties in the formulation and use of the substances.
6. Impurities may bring about an incompatibility with other substances.
7. Impurities may lower the shelf life of the substance.
8. Impurities, though harmless in nature, may bring about changes in odour, colour, taste, etc. thus making the use of the substance unhygienic.
199. How to determine the Permissible Impurities’ limit in Pharmaceutical Substances?
Following points are considered while determining impurity limits.
1. For impurities which are of harmful type such as lead, arsenic etc., a low permissible limit is prescribed. This is based upon, how much of these can be tolerated? Which itself is based upon how much of the impurity is harmful?
2. For impurities that are harmless, the aim is to fix their limits so that their presence does not interfere in the therapeutic usefulness of the drug. Here, again, the limits are prescribed and fixed. This is done depending upon the nature of the impurity, the type of substance, use of the substance etc.
3. Practicability of obtaining substances without impurities at reasonable costs. It may be possible to prepare substances through a series of steps of purification without any impurities, but this could escalate the cost. Considering this aspect, limits of various impurities are fixed.
4. Deliberate adulteration by using materials having similar qualities also accounts for the presence of impurities in the substance, e.g. adulteration of sodium salt with potassium salt, calcium salts with magnesium salts etc. Such adulteration which brings impurities into substances, need not exhibit less therapeutic activity but it is reasonable to expect unadulterated material from an ethical point of view. Pharmacopoeias guard against this type of impurity by employing tests for identification.
200. What are the regulatory requirements for the management of impurity?
Following are the regulatory requirements for the management of impurity

Inspired by: https://www.intechopen.com/
201. Explain the classification of impurities.
Impurities are mainly classified as Organic, Solvents and Inorganic.Details are as follows:

202. What are the general tests for detecting impurities in the pharmaceuticals.
1. Colour, Odour and Taste
2. Physico-chemical Constants
3. Acidity, Alkalinity and pH
4. Anions and Cations
5. Insoluble Residue
6. Ash, Water Insoluble Ash
Limit tests in the pharmaceuticals
203. What are the limit tests in pharmaceuticals?
Limit tests are defined as quantitative or semi-quantitative tests which are performed to identify and control small quantities of impurities which are likely to be present with the substances to be analyzed.
204. Explain the Limit Test for Chloride?
The limit test for chlorides is based upon the chemical reaction between soluble chloride ions with a silver nitrate reagent in a nitric acid media. The insoluble silver chloride renders the test solution turbid (depending upon the amount of silver chloride formed and therefore, on the amount of chloride present in the substance under test.) This turbidity is compared with the standard turbidity produced by the addition of silver nitrate, to the known amount of chloride ion (sodium chloride) solution. If the test solution shows less turbidity than the standard, the sample passes the test.
Dissolve the specified quantity of substance in water or prepare a solution as directed in the pharmacopoeia and transfer to a Nessler’s cylinder A. Add 1 ml of dilute nitric acid except when nitric acid is used in the preparation of the solution. Dilute it to 50 ml with water and add 1 ml of silver nitrate solution, stir immediately with a glass rod and set aside for 5 minutes.
Simultaneously for standard opalescence, place 1 ml of 0.05845 per cent w/v solution of sodium chloride in Nessler’s cylinder B and add 10 ml of dilute nitric acid, make up the volume to 50 ml with water, add 1 ml of silver nitrate solution, stir with the glass rod and set aside for 05 minutes. The opalescence produced by the sample (in cylinder A) should not be greater than standard opalescence.
205. Explain the Limit Test for Sulphate?
In a limit test for sulphate, the solution of the substance under test is mixed with barium sulphate reagent in a hydrochloric acid medium and the turbidity so produced is compared with the standard in similar manner with a known quantity of sulphate ion (using potassium sulphate). The substance passes the limit test if it produces a turbidity that is less than the standard.
A solution of specified quantity of substance is made in water or prepared as directed in the pharmacopoeia in Nessler’s cylinder; add 2 ml dilute hydrochloric acid except where hydrochloric acid is used in the preparation of solution. Dilute it to 45 ml with water, add 5 ml of barium sulphate reagent, stir immediately with the glass rod and set aside for 5 minutes. To produce standard turbidity, place 1 ml of 0.1089 percent w/v solution of potassium sulphate and 2 ml of dilute hydrochloric acid in another Nessler’s cylinder, dilute to 45 ml with water, add 15 ml of barium sulphate reagent, stir immediately and set aside for 5 minutes. The turbidity produced by the sample solution is not greater than the standard turbidity.
British Pharmacopoeia makes use of a barium sulphate reagent, which contains barium chloride, alcohol and small amounts of potassium sulphate. Alcohol prevents super-sa turation, and potassium sulphate increases the sensitivity of the test by giving the ionic concentration in the reagent, which just exceeds the solubility product of barium sulphate.
206. Explain the Limit Test for Iron?
This test is based upon the reaction of iron in an ammoniacal solution, with thioglycollic acid which forms a pink to deep reddish purple coloured complex of iron -thioglycoliate. The colour produced from a specified amount of substance from the test, is compared by viewing vertically, with a standard (Ferritic ammonium sulphate). If the colour from test solution is less dark than the standard then the sample passes the test.
The Fe (SCH2 COOH)2 formed with the ferrous form of iron, is quite stable for long period in the absence of air. The colour, however, is destroyed by oxidizing agents and strong alkali s. The original state of iron is unimportant, as thioglycollic acid reduces Fe3+ to Fe2+.
This test is very sensitive. Interference of other metal cations is eliminated, by making use of 20 percent citric acid, which forms complex with other metal cations.
Prepare a solution by dissolving a specified amount of substance in 40 ml water or take 10 ml of solution as directed in the monograph in Nessler’s cylinder. Add 2 ml of 20 per cent w/v solution of iron free citric acid and 0.1 ml thioglycollic acid, mix and make alkaline with iron free ammonia solution and dilute it to 50 ml with water. Allow to stand for 5 minutes. For standard, imultaneously dilute 2 ml of standard iron solution with 40 ml of water, add the same quantity of reagent as in the sample. Any colour produced by the sample is not more intense than the standard.
Earlier, ammonium thiocyanate reagent was used for the limit test of iron. Since thioglycollic acid is more sensitive reagent for iron, it has replaced ammonium thiocyanate in the test.
207. Explain the Limit Test for Heavy Metals?
The Indian Pharmacopoeia adopts three methods for the limit tests for heavy metals. The ‘Method A’ is used for the substance which yields a clear colourless solution under specified conditions. ‘Method 8’ is used for those substances which do not yield clear colourless solution under the test conditions specified for method A. ‘Method C is used for substances that yield clear colourless solution in sodium hydroxide medium. The reagents like acetic acid, ammonia, hydrochloric acid, nitric acid, potassium cyanide and sulphuric acid should be lead free and designated as ‘Specific reagents ‘.
Method A:
Standard solution is prepared by taking 2 ml of standard lead solution and by diluting it to 25 ml with water. The pH is adjusted between 3 and 4 by using either dilute acetic acid or dilute ammonia solution. Make up the volume of 35 ml with water.
Test solution is prepared as directed in the individual monograph. Take 25 ml and adjust the pH of the solution between 3.0 and 4.0 by using dilute acetic acid or dilute ammonia and adjust the volume to 35 ml with water.
To each of the cylinders containing standard and test solution, add 10 ml of freshly prepared hydrogen sulphide solution, mix, dilute to 50 ml with water and allow it to stand for 5 minutes. The colour when viewed downwards over white surface shou ld not be darker for the test than the standard solution.
Method B:
The standard solution is prepared as directed under method A. Test solution is prepared in a crucible by weighing a specified quantity of substance as per monograph. Moisten the substance with sulphuric acid, ignite on a low flame till completely charred. Add few drops of nitric acid and heat to SOOT Allow to cool, add 1 ml of hydrochloric acid and evaporate to dryness. Moisten the residue with 10 ml hydrochloric acid and digest for two minutes.
Neutralize with ammonia solution and make just acidic with acetic acid. Adjust the pH between 3.0 and 4.0, filter if necessary. Adjust the volume of filtrate to 35 ml in Nessler’s cylinder, add 10 ml of hydrogen sulphide solution, dilute to 50 ml with water and compare the colour with standard solution.
Method C:
The standard solution is prepared by using 2 ml of standard lead solution; adding 5 ml dilute sodium hydroxide solution and making the volume to 50 ml with water. For the test solution, take either 25 ml solution prepared as directed in the monograph or dissolve specified quantity of substance in 20 ml water, add 5 ml of dilute sodium hydroxide solution and make up the volume to 50 ml.
To each of the above solution in Nessler’s cylinder add 5 drops of sodium sulphide solution, mix and set aside for 05 minutes. The colour produced by test solution should not be darker than the standard solution.
208. Explain the Limit Test for Volatile Oils?
In 25 ml glass stoppered test tubes; 10 ml of the oil is mixed with an equal volume of water containing a drop of hydrochloric acid. Hydrogen sulphide is passed through the mixture until it is saturated. No darkening in colour should be produced neither in the oil, nor in the water layer, for the sample to pass the test.
209. Explain the Limit Test for Lead?
The limit test for lead as per IP and USP is based upon the reaction between lead and diphenylthiocarbozone (dithizone). Dithizone in chloroform extracts lead from alkaline aqueous solutions as a lead Dithizone complex (red in colour).
The original dithizone has a green colour in chloroform thus the lead-dithizone shows a violet colour. The intensity of the colour of the complex depends upon the amount of lead in the solution. The colour of the lead-dithizone complex in chloroform is compared with a standard volume of lead solution, treated in the same manner.
In this method, the lead present as an impurity in the substances, is separated by extracting an alkaline solution with a dithizone extraction solution. The interference and influence of other metal ions etc. is eliminated by adjusting the optimum pH for the extraction, by using ammonium citrate, potassium cyanide, hydroxylamine hydrochloride reagents etc.
Method:
A known quantity of the sample solution is taken in separating funnel: 6 ml of ammonium citrate, and 2 ml of hydroxylamine hydrochloride is added, followed by 2 drops of phenol red, and the solution is made alkaline by adding an ammonia solution. Add 2 ml of potassium cyanide solution and extract immediately with 5 ml portions of dithizone solution (till green). The combined dithizone extracts are shaken for 30 seconds, with 30 ml of 1 percent nitric acid, and the chloroform layer discarded. To the acid solution 5 ml standard dithizone solution is added along with 4 ml of ammonium cyanide and shaken for 30 seconds. A known quantity of the standard solution of lead (equivalent to the amount of lead permitted in the sample) is treated separately. The violet colour of the chloroform layer- of the sample- should not be darker than the standard for the sample to pass the test.
In the preparation, an appropriate preliminary treatment is given to get lead in the solution, without any interfering substance or ion. All reagents employed under the test (except for standard lead solution), should be free from lead, and are designated as ‘PbT’ reagents in pharmacopoeias.
Limit Test for Lead as per British Pharmacopeia:
British Pharmacopeia adopts another method for the limit test for lead which is based on the formation of a brownish colouration produced by the colloidal lead sulphide upon addition of sodium sulphide to the solution under test. If the lead content is more, then a brownish black precipitate of lead sulphide is obtained. The colour produced in the test solution is matched against the standard that is made from a known amount of lead in a Nessler’s cylinder. In order to carry out this test two solutions, a primary and an auxiliary are prepared from the sample.
Method:
Two solutions of the substance under test are prepared with hot water and acetic acid. One is the primary solution containing a definite but greater amount of substance and placed in a 50 ml Nessler’s cylinder. The other is the auxiliary solution containing a known amount of the test substance in another 50 ml Nessler’s cylinder. To this auxiliary solution, a definite amount of a dilute solution of lead nitrate is added. Ammonia and potassium cyanide solutions are added to both the solutions in the Nessler’s cylinders. Small amounts of burnt sugar solution are added to both solutions; to correct any difference of colour and the volume is made up to 50 ml. If the solutions appear turbid, they are filtered and the volume made up to SO ml. Both solutions are treated with sodium sulphide solution and a colour is developed. If the colour in the auxiliary solution is darker than that in the primary, the substance contains lead within limits.
The object of using primary and auxiliary solutions of substances is to have a comparison made under identical conditions. Interference by any unknown entity present in the solution is eliminated by this technique.
210. Explain the Limit Test for Arsenic?
Arsenic is an undesirable and harmful impurity in medicinal substances, and all pharmacopoeias prescribe a limit test for it. There are many qualitative and quantitative tests for arsenic. The pharmacopoeial method is based on the Gutzeit test. In this test, arsenic is converted into arsine gas, (AsH3) which when passed over a mercuric chloride test paper, produces a yellow stain. The intensity of the stain is proportional to the amount of arsenic present. A standard stain produced from a definite amount of arsenic is used for comparison.
The chemical reactions involved in the method are given below:
When the sample is dissolved in acid, the arsenic present in the sample is converted to arsenic acid. The arsenic acid is reduced by reducing agents like potassium iodide, stannous chloride etc. to arsenious acid.
The nascent hydrogen produced during the reaction, further reduces arsenious acid to arsine (gas), which reacts with mercuric chloride paper, producing a yellow stain.
To carry out the test, a specified apparatus (as described in pharmacopoeias) is used. In order to convert arsenic into arsine gas, various reducing agents like zinc and hydrochloric acid, stannous chloride and potassium iodide are employed. The rate of evolution of gas is maintained by using a particular size of zinc, and controlling the concentration of acids and
other salts of the reaction medium, besides temperature. Any impurity coming along with the gas (like H,S) is trapped by placing a lead acetate soaked cotton plug in the apparatus. All the reagents employed for the test should be arsenic-free, and are designated as AsT in pharmacopoeias.
Electrochemical methods of analysis in the pharmaceuticals
211. Which are the electrochemical methods of analysis?
• Conductometry
• Potentiemetry
• Polarography
212. What is Conductometry analysis?
Conductometry involves determination of conductance of an electrolyte solution using a conductometer. When solutions of electrolytes are subjected to an electric field, they conduct electric current by migration of ions. This process of conductance obey Ohm’s law.
Conductance is expressed as the reciprocal of resistance i.e. 1/R and it is denoted by unit
known as mhos or ohms-I.
There are different types of conductance as follows:
1. Specific conductance
2. Equivalent conductance
3. Molar conductance
213. What are the applications of conductometry analysis?
- Conductivity meter
- Conductometric Titrations
- Strong acid with strong base
- Strong acid with weak base
- Weak acid with strong base
- Weak acid with weak base
- Very weak acid with strong base
- Mixture of hydrochloric acid (strong acid) and acetic acid (weak acid) with strong base
- Displacement titrations
- Precipitation and complex formation titrations
- Redox titrations
- Determination of solubility of sparingly soluble material
- Kinetic studies
- Degree of dissociation of weaker electrolytes
- Basicity of organic acids
- Determination of concentration:
214. What is potentiometry analysis?
Different electrode systems are used in combination to measure potential or pH (hydrogen ion concentration) of a solution. A pair of electrode is commonly required in measurement. One electrode acts as an indicator electrode, while the other as reference electrode.
215. Example of potentiometry reference electrodes, its advantages and disadvantages?
A. Normal Hydrogen Electrode (NHE)
- Advantages of Hydrogen Electrode (NHE)
- It is a fundamental electrode and is used as a standard in pH measurements.
- It can be used over a wide pH range.
- It exhibits no salt error.
- It establishes equilibrium rapidly and gives accurate results.
- Disadvantages of Hydrogen Electrode
- It can not be used in solutions containing strong oxidizing or reducing agents.
- It can not be used in solutions containing metal ions that are below hydrogen in
- potential series (In such cases interaction with hydrogen will occur and the metal
- will be deposited on the electrode surface).
- It gets readily poisoned by a number of substances like proteins, tannins, mercury salts etc.
- It is cumbrous to prepare, and use in routine analysis.
B. Calomel electrode
- Bell jar type
- Side arm test-tube type
- Test-tube type
- Advantages of Calomel Electrode are:
- It is sturdy and useful for measurement of a wide pH range.
- It can be employed in various solvents.
- Disadvantages of Calomel Electrode are:
- It is unstable at higher (above 80°) temperature and
- It is unsuitable where chloride ions show incompatibility.
C. Silver/ Silver Chloride Electrode
D. Mercury-Mercurous Sulphate Electrode
216. Example of indicator electrodes, its advantages and disadvantages?
1. Hydrogen electrode
2. Quinhydrone electrode
- The advantage of this electrode is that it attains equilibrium rapidly and can be used where the hydrogen electrode is unsuitable. It gives accurate results also.
- Disadvantage of this electrode is that it can not be used in more alkaline solutions whose pH is above 8 as it readily gets oxidized by air in alkaline medium.
3. Antimony electrode
Advantages of antimony electrode include:
- It can be used for measuring pH ranging from 2 to 8.
- It can be used in viscous or turbid solvents.
- It is sturdy and therefore, it is very useful where continuous pH recordings are made.
- The electrode does not get readily poisoned.
The disadvantages of antimony electrode are:
- It can not be used in measuring pH below 03 as the oxide gets dissolved.
- It can not be used in presence of strong oxidizing agents and complexing agents.
- It can not be used in presence of metals such as Cu, Ag, Au which are more noble than antimony (i.e. below in electromotive series).
- It suffers from salt error. A modified micro antimony electrode is currently used in industry.
4. Glass electrode
Advantages of glass electrode:
- It exhibits reasonably rapid response over a wide range of pH.
- It is uninfluenced by the presence of oxidizing or reducing agents.
- It can be used in viscous, coloured solutions, suspensions of colloidal solutions.
Disadvantages of glass electrode:
- It is fragile and hence should be handled carefully – minute scratches render the electrode useless.
- It is unsatisfactory in more alkaline, above pH 10 range, as there is a partial exchange of other cations than hydrogen ion through membrane.
Karl fischer method for determination of water
217. What is the purpose of the Karl fischer method of analysis?
The method of analysis proposed by Karl Fischer (in 1935) is considered as relatively specific for water. It essentially makes use of the Karl Fischer reagent which is composed of iodine, sulphur dioxide, pyridine and methanol. Pyridine and methanol used during the method of analysis should be anhydrous.
218. Explain the theory of the Karl fischer method of analysis?
Water present in the analyte reacts with the Karl Fischer reagent as per following two-stage process:
Stage 1:

Stage 2:

Oxidation of sulphur dioxide takes place by iodine to yield sulphur trioxide and hydrogen iodide thereby consuming one mole of water. Each one molecule of iodine disappears against each molecule of water present in the given sample. It is pertinent to mention here that in the presence of a large excess of pyridine (C5H5N), all reactants as well as the resulting products of reaction mostly exist as complexes as evident from above equations.
219. Explain the precautions while using the Karl Fischer reagent.
- Always prepare the reagent a day or two before it is to be used,
- Great care must be taken to prevent and check any possible contamination either of the reagent or the sample by atmospheric moisture,
- All glassware(s) must be thoroughly dried before use,
- Standard solution should be stored out of contact with air, and
- Essential to minimize contact between the atmosphere and the solution during the course of titration.
Optical method of analysis
220. What are the examples of optical methods of analysis?
- Refractometry
- Polarimetry
- Nephelometry
- Turbidimetry
221. What is the Refractometry method of analysis?
Light passes more rapidly through a vacuum than through a substance (medium). It has been observed that when a ray of light happens to pass from one medium (a) into another medium (b) it is subjected to refraction. The ray travels at a lower velocity in the relatively more optically dense medium (b) than in medium (a) which is less optically dense.

Diagram represents the Path of Light between Two Media ‘a’ and ‘b’
222. What are the various applications of refractivity?
(a) Determine the molar refractivity of different substances and comparing their values with theoretical ones
(b) Molar refractivity is an additive property, hence, it may be utilized to determine the refractivities of homogeneous mixtures (as solutions).
(c) Determination of Critical Micelle Concentration.
223. What is the polarimetry method of analysis?
An optically active substance is one that rotates the plane of polarized light. In other words, when a polarized light, oscillating in a specific plane, is made to pass through an optically active substance, it happens to emerge oscillating in an altogether different plane.
224. Explain the principle of a polarimeter.
Principle of a polarimeter is that light from the source, usually a sodium vapour lamp, first gets collimated at A, and subsequently falls upon polarizer B (a calcite prism). The polarizer
permits only the light polarized in a particular direction to pass it. The emergent polarized ray now passes through the sample under investigation, kept in the polarimeter glass tube C to the analyzer D, which happens to be another polarizing prism. The analyzing rotator prism D (Nicol) is fixed in such a manner that it can be rotated easily about the axis of the incident light ray. Two situations arise when the analyzing rotator prism (D) is put into action, firstly, the prism being parallel to the plane of polarization of the incident light—the net result is that the intensity of light reaching the Null detector F is maximum ; and secondly, the prism being perpendicular
to the plane of the polarized light—the net result is observed by the intensity of light reaching the detector as minimum. Hence, the overall difference in the position of the analyzer, as noted from the circular scale E, that provides minimum light intensity with and without the sample in the cell is the observed ‘rotation’ of the sample in question.

A = Collimated monochromatic light source,
B = Polarizing prism (Nicol),
C = Polarimeter glass tube (20 cm) with glass windows,
D = Analyzing rotator prism (Nicol),
E = Circular scale with vernier,
F = Null detector (Eye or Photoelectric Cell).
225. Explain Specific Rotation.
A polarized light when passed through an optically active substance, each molecule of it encountered by the light beam rotates the plane of polarization by a constant amount characteristic of the substance. Consequently, a measure of the rotary power of the individual molecule, irrespective of the two parameters, namely : the path length and the concentration, is achieved by converting the measured rotation into a specific rotation.
226. Explain Turbidimetry.
When light is passed through moderately stable suspensions, a portion of the incident radiant energy is dissipated by virtue of the absorption, refraction, and reflection, whereas the remaining portion gets transmitted. It is quite evident that the optical characteristics of each suspension shall alter according to the concentration of the dispersed phase. The measurement of the intensity of the transmitted light through such suspensions vis-a-vis the concentration of the dispersed phase serves as the basis of turbidimetric analysis.
In short, turbidimetry is the measurement of the degree of attenuation of a radiant beam incident on particles suspended in a medium, the measurement being made in the directly transmitted beam.
227. Explain Nephelometry.
When light is passed through moderately stable suspensions, and it is viewed at 90° (i.e., right angles) to the direction of the incident light the system appears opalescent on account of the reflection of light from the particle of the suspension. This scattering of light is termed as the Tyndall effect.
The observed opalescence or cloudiness is the net result caused by irregularly and diffusely reflected light from the suspension. Consequently, the ultimate measurement of the intensity of the scattered light as a true representation of the actual concentration of the dispersed phase forms the basis of nephelometric analysis (derived from Greek : nephele-means cloud).
It is found to be most sensitive and effective specially in the case of very dilute suspensions having a concentration not greater than 100 mg L–1. However, it is interesting to observe that the technique of turbidimetric analysis resembles that of flame photometry ; and nephelometric analysis to that of fluorimetry.
Nephelometry exclusively refers to the measurement of the light scattered by suspended particles at right angles (perpendicular) to the incident beam.
Nuclear Magnetic Resonance (NMR) spectroscopy
228. Explain the Nuclear Magnetic Resonance (NMR) spectroscopy.
In Nuclear Magnetic Resonance (NMR) spectroscopy, energy from an external source is absorbed and brings about a change or resonance to an ‘excited’ or high energy state.
The energy required for NMR lies in the low energy or long wavelength radio-frequency end of the electromagnetic spectrum. Because of the magnetic properties of nuclei arising from the axial spin, the emerging radiofrequency gets absorbed in a magnetic field.
Because of this, for a particular nucleus an NMR absorption spectrum invariably comprises one to several groups of absorption lines in the ratio-frequency portion of the electromagnetic spectrum.
Hence, the location of peaks indicate the chemical nature of the nucleus, whereas the multiplets provide information regarding the spatial positions of the neighbouring nuclei. For this reason the NMR is also known as Nuclear Spin Resonance (NSR) spectroscopy.
Therefore, NMR spectroscopy finds its applications for compound identification, by means of a ‘fingerprint technique’ very much identical to that used in IR-spectroscopy. Besides, it is invariably utilized as a specific method of assay for the individual constituents of a mixture. A few typical examples of drug assays will be dealt separately at the end of this chapter to justify its efficacy and usefulness.
229. Explain the NMR phenomenon.
A. The Spinning Nucleus : The nucleus of the hydrogen atom, i.e., the proton, just behaves as if it is a small spinning bar magnet. It does so because it evidently possesses an electrical charge as well as a mechanical spin. Consequently, a spinning charged body will generate a magnetic field, and hence the nucleus of hydrogen atom is not an exception
B. The effect of an External Magnetic Field : As a ‘compass needle’ possesses an inherent tendency to align itself with the earth’s magnetic field, the proton not only responds to the influence of an external magnetic field but also tends to align itself with that field. However, because of restrictions as applicable to nuclei (not to compass needles) the proton can only adopt the following two orientations with regard to an external magnetic field.
(a) when proton is aligned with the field (i.e., at lower energy state),
(b) when proton is opposed to the field (i.e., at higher energy state).
C. The Precessional Motion: The proton appears to be behaving as ‘spinning magnet’ and therefore, not only can it align itself with or oppose an external field, but also may move in a characteristic manner under the influence of the external magnet.
D. The Precessional Frequency: The spinning frequency of the nucleus does not change at all, whereas the speed of precession does. Therefore the precessional frequency is directly proportional to the strength of the external field. It designates one of the most important relationships in NMR-spectroscopy.
E. The Energy Transitions: Whenever a proton is precessing in the aligned orientation (low energy) it can absorb energy and pass into the orientation (high energy); and subsequently it can lose this extra energy and relax back into the aligned state.
230. What information is provided by 1H-NMR (PROTON-NMR)?
1H-NMR provides many valuable information that are used for the structural elucidation as well as assay of important pharmaceutical substances. Details are as follows:
(i) To record differences in the magnetic properties of the various nuclei present,
(ii) To deduce in large measure the exact locations of these nuclei within the molecule,
(iii) To deduce how many different types of hydrogen environments are present in the molecule,
(iv) To deduce which hydrogen atoms are present on neighbouring carbon atoms, and
(v) To measure exactly how many H-atoms are actually present in each of these environments.
231. Explain 3H-NMR (TRITIUM NMR-SPECTROSCOPY).
The ease with which ‘tritium’ could be employed for labelling organic compounds, having fairly high molar specific activity, has turned it into a very useful and versatile β-emitting radionuclide for chemical and life sciences research.
The unique novel characteristic feature of tritium tracers being that it may be used as a tracer for carbon as well as hydrogen structures.
232. Advantages of 3H-NMR (TRITIUM NMR-SPECTROSCOPY).
• A rapid direct and non-destructive method,
• Provides direct information on regiospecificity,
• Gives quantitative distribution of the label,
• Caters for accurate and precise information on the stereochemistry of the label, and
• Requires only millicuries rather than microcuries or lesser amounts of radioactivity.
233. Explain 13C-NMR SPECTROSCOPY.
The ‘carbon-skeleton’ has been viewed directly with the help of Carbon-13 NMR spectroscopy on a particle basis since the early 1970’s; whereas 1H-NMR spectrometry started in the late 1950’s. The valuable contribution made by various researchers, between 1976 and 1980, has virtually placed 13C-NMR to a strategically much advanced stage where it gives a clear edge over 1H-NMR in terms of not only its versatility but also its wide application in analysis.
13C-NMR refers to recording another NMR-spectrum but of the C-13 atoms rather than the hydrogen atoms. In actual practice, however, -‘these spectra are recorded in such a manner that each chemically distinct carbon gives rise to single peak, without any coupling or fine structure’.
Hence, simply a count of the peaks can be used to see how many carbons are actually present in the molecule. But this particular technique is not reliable for a molecule that exhibits symmetry, because this would ultimately reduce the number of peaks.
234. Explain 2D-NMR (TWO DIMENSIONAL CORRELATION SPECTROSCOPY OR TWO DIMENSIONAL COSY SPECTRUM)
The interaction between different hydrogens in a molecule, known as ‘scaler’ or ‘spin-spin coupling’, transmitted invariably through chemical bonds, usually cover 2 or 3 at the most. Therefore, when a hydrogen with a chemical shift ‘A’ is coupled to a hydrogen with chemical shift ‘B’, one would immediately make out that the hydrogens must be only 2 or 3 bonds away from one another. To know exactly with particular hydrogens are coupled to one another it is necessary to record a two-dimensional ‘Correlation Spectroscopy’ (COSY) spectrum.
Generally, a normal NMR-spectrum has amplitude plotted Vs just one frequency-dimension (the ppm scale). In 2D-NMR, the amplitude is plotted Vs two frequency-dimensions (two ppm scales), normally in the form of a counter plot, just like a topographic map.
The most important aspect about these 2D-NMR spectra is that they show the relation between the peaks in an NMR-spectrum.
235. Applications of NMR-spectroscopy in pharmaceutical analysis
a. Identification testing
b. Assay of drugs
Emission spectroscopy
236. Explain Emission spectroscopy.
In emission spectroscopy, the atoms present in a sample undergo excitation due to the absorption of either electrical or thermal energy. Subsequently, the radiation emitted by atoms in an excited sample is studied in an elaborated manner both qualitatively and quantitatively.
237. Use of emission spectroscopy.
Emission spectroscopy is useful analytical tool for the analysis of :
- Elemental analysis of metals,
- Identification and quantitative determination of metallic elements,
- Estimation of metalloids e.g., arsenic, silicon, selenium, present is extremely low concentrations,
- Analysis of solids, liquids or gases as follows :
- solids-as such or evaporated solutions,
- liquids-atomized spray, analyzed occasionally, and
- gases-analyzed rarely.
238. Explain the theory of emission spectroscopy.
Emission spectroscopy can be explained using following aspects:
(a) Spectra: A beam of light on being passed either through a Nicol’s prism or a grating, is split-up right into its constituent array of colours frequently termed as spectrum. However, the complete spectrum has a wide range that may be further divided into various regions based on their respective wavelengths (0 to 35,000° A), i.e Ultraviolet Region, Visible Region, and Infrared Region.
(b) Classes of Spectra: There exist, in fact, two major types of spectra commonly termed as emission spectra and the absorption spectra.
(c) Classification of Emission Spectra: The emission spectra may be classified into the following three types, namely, i. Band Spectra (or Molecular Spectrum), ii. Continuous Spectra, and iii. Line Spectra Effect of Concentration on Line and Band Spectra : The radiant power by virtue of the radiant energy, of a line or band exclusively depends directly on the total number of excited atoms or molecules present, which is subsequently proportional to the total concentration of the species present in the source.
(d) Effect of Concentration on Line and Band Spectra: The radiant power by virtue of the radiant energy, of a line or band exclusively depends directly on the total number of excited atoms or molecules present, which is subsequently proportional to the total concentration of the species present in the source.
(e) Excitation-Energy Requirements: A single spectral-line is emitted from an element only when the energy equivalent to the excitation potential of the element is usually absorbed. This particular requirement is very critical and important. Exactly in a similar manner, the full-fledged complete spectrum is obtained possibly only when the energy equivalent to the ionization potential is absorbed by a molecule.
239. Explain the Salient Features of Excitation Sources.
Salient Features of Excitation Sources are follows:
• Sample should be changed into its vaporized form,
• Vaporized form of sample must be dissociated into atoms,
• Electrons present in the atoms should be excited from the ground state to higher-energy levels,
• Capable of exciting atoms of most of the elements of interest (in the Periodic Table),
• To produce sufficient line-intensity in order to detect these lines within the scope of the ‘detection limit’, and
• Must essentially achieve reproducible excitation conditions of various samples.
240. Explain the types of Excitation Sources.
(i) Flames
(ii) Direct Current Arc
(iii) Alternating Current Arc
241. Explain the types of electrodes.
(a) Self Electrodes
(b) Graphite Electrodes
242. Explain the process of sample handling in emission spectroscopy.
(a) Solids: Solid samples can also be sub-divided into two categories, such as : (i) Those possessing good conductance characteristics and can withstand high temperatures : it can be achieved by making electrodes with the material directly to be used for the electrical discharge ; (ii) Those having poor conductance and cannot withstand high temperatures : it can be powdered mixed with the powdered graphite (known as buffer) and placed in the depression of the lower graphite electrode. On passing the electrical discharge the material (sample) is first vaporised into the body of the discharge and subsequently the spectrographic emission occurs.
(b) Liquids: Liquid samples may be dispensed conveniently with the aid of two types of smallholders, namely : firstly, wherein the porous base of the cup gradually releases the sample into the discharge from the top ; and secondly, wherein the rotating-disc carriers take up the sample into the discharge from the bottom steadily.
243. Which monochromators are used in emission spectroscopy?
(a) Prism Monochromators
(b) Grating Monochromators
244. Which Detectors are used in emission spectroscopy?
(a) Photographic Detectors-used for qualitative analysis,
(b) Photomultiplier Detectors-used for quantitative analysis
245. Explain the applications of emission spectroscopy.
1. Emission spectroscopy is used for the analysis of various alloys, namely : aluminium, copper, magnesium, zinc, lead, and tin.
2. It is used for the analysis of a number of elements, for instance : Na, K, Zn, Cu, Ca, Mg, Ni and Fe present in various tissues of human beings. Changes in trace-metal concentrations can be studied at length with regard to the ageing process.
3. Trace amounts of Ca, Cu, and Zn can be examined in blood samples.
4. Presence of Zn can be examined in the pancreas tissue.
5. To determine the extent of elements present in ‘crude oil’ by virtue of the fact that some of these may poison the catalysts used in the cracking-process e.g., V, Cu, Ni, and Fe.
Flame Emission Spectroscopy (FES)
246. What is Flame Emission Spectroscopy (FES)?
Metallic salts or metallic compounds after dissolution in appropriate solvents when introduced into a flame (for instance : acetylene burning in oxygen at 3200°C), turns into its vapours that essentially contain mostly the atoms of the metal.
Such gaseous metal atoms are raised to a particular high energy level that enables them to allow the emission of radiation characteristics features of the metal. (Example: the characteristic flame colourations of metals frequently encountered in simple organic compounds such as : Na-yellow, Ca-brick-red ; Ba-apple-green) This forms the fundamental basis of initially called Flame Photometry, but more recently known as Flame Emission Spectroscopy (FES).
It is evident that a relatively large proportion of the gaseous metal atoms shall remain in the ground state i.e., in an unexcited form. It has been observed that such ground-state atoms shall absorb radiant energy pertaining to their own particular resource wavelength. Therefore, when a light having the same resonance wavelength is made to pass through a flame consisting of such atoms, a portion of the light shall be absorbed accordingly.
The extent or degree of absorption would be directly proportional to the total number of ground-state present in the flame.
247. Explain the steps of Flame Emission Spectroscopy (FES) and Atomic Absorption Spectroscopy (AAS)
The emission spectrum obtained is made up of a number of lines that actually originate from the resulting excited atoms or ions ; and these steps are explained as follows.
Step-1: The liquid sample containing a suitable compound of the metal (M+ A–) is aspirated into a flame, thereby converting it into its vapours or liquid droplets,
Step-2: The evaporation of vapours (or droplets) give rise to the corresponding solid residue,
Step-3: The vapourization of the solid residue into its gaseous state occurs,
Step-4: The dissociation of the gaseous state into its constituent atoms, namely : M(gas)+ A(gas) take place, that initially, is in ground state,
Step-5: The thermal excitation of some atoms into their respective higher energy levels will lead ultimately to a condition whereby they radiate energy (flame emission) measured by Flame Emission Spectroscopy (FES), and
Step-6: The absorption of radiant energy by some atoms into their higher energy levels enable them to radiate energy (atomic absorption) measured by Atomic Absorption Spectroscopy (AAS).
248. What is the principle of Flame Emission Spectroscopy (FES)?
The principle of Flame Emission Spectroscopy (FES) can be explained when a liquid sample containing a metallic salt solution under investigation is introduced into a flame, the following steps normally takes place in quick succession, namely :
(i) the solvent gets evaporated leaving behind the corresponding solid salt,
(ii) the solid salt undergoes vaporization and gets converted into its respective gaseous state,
(iii) the progressive dissociation of either a portion or all of the gaseous molecules gives rise to free neutral atoms or radicals.
The resulting neutral atoms are excited by the thermal energy of the flame which are fairly unstable, and hence instantly emit photons and eventually return to the ground state (i.e., the lower energy state). The resulting emission spectrum caused by the emitted photons and its subsequent measurement forms the fundamental basis of FES.
249. Explain Types of Flame Photometers.
There are two types of Flame Photometers that are used invariably in Flame Emission Spectroscopy (FES) which are as follows :
(a) Simple Flame Photometer
(b) Internal Standard Flame Photometer
250. Explain the applications of flame emission spectroscopy in pharmaceutical analysis.
i. Assay of sodium, potassium and calcium in blood serum and water
ii. Assay of barium, potassium and sodium in calcium acetate
ii. Cognate assays
Atomic Absorption Spectrophotometer
251. Explain Atomic Absorption Spectrophotometer.
In atomic absorption spectroscopy (AAS) the sample solution is aspirated directly into a flame or by using an electrothermal device-whereby the sample solution is first evaporated and then ignited on a hot surface. It has been noticed that gaseous metal atoms in an unexcited form i.e., ground state atoms, will absorb radiant energy related to their own specific resonance wavelength. Hence, when a light with the same resonance wavelength is passed through a flame comprising of such atoms, a part of the light will be absorbed accordingly. Besides, the degree of absorption would be directly proportional to the total number of ground-state atoms present in the flame, which ultimately forms the basis of Atomic Absorption Spectroscopy (AAS).
252. Explain the theory/ principle of Atomic Absorption Spectrophotometer.
The principle of atomic absorption spectroscopy (AAS) is the absorption of energy exclusively by ground state atoms while they are in the gaseous form.
A solution consisting of certain metallic species when aspirated into a flame, it will give rise to the corresponding vapours of metallic species. As it has already been discussed under flame emission spectroscopy (FES): Some metal atoms would be raised directly to an energy level to such an extent as to emit the particular radiation of the metal. At this critical point, a sufficiently large quantum of the metal atoms of a particular element would still remain in the non-emitting ground-state, which in turn shall be receptive of light radiation having their own specific wavelength. Consequently, when a light of this wavelength is passed through a flame ; along the atoms of the metallic species, a portion of the same would be absorbed; and the resulting absorption has been found to be directly proportional to the density of the atoms present in the flame at that material time. The concentration of the metallic element may be determined directly from the value of absorption.
253. Explain advantages of Atomic Absorption Spectrophotometer (AAS) over Flame Emission Spectroscopy (FES).
| Atomic Absorption Spectrophotometer (AAS) | Flame Emission Spectroscopy (FES) |
| This technique is superior and specific because of the fact that only the atoms of a particular element can absorb radiation of their own characteristic wavelength. | Spectral interferences usually take place in this technique. |
| A relatively large number of metal atoms produce an atomic absorption signal whereby the effect of flame-temperature variation is negligible in AAS i.e., independent of flame-temperature. | A much smaller number of metal atoms do produce an emission signal in FES, showing that this technique is not independent of flame, temperature. |
| The detection limits of sensitivity of the following elements are more by AAS technique, such as : Ag, As, Au, B, Bi, Cd, Co, and Fe. | The detection limits (sensitivity) of the undermentioned elements are higher by FES technique, for instance: Al, Ba, Ca, Eu, Ho, In, K and La. |
254. Disadvantages of Atomic Absorption Spectrophotometer (AAS).
The various points of demerit of atomic absorption spectroscopy are as follows :
(1) It essentially requires a separate lamp for each element to be determined ; and this serious lacuna is usually overcome either by using a line-source with the introduction of flame or by using a continuous source with the introduction of a very high resolution monochromator,
(2) AAS cannot be employed very effectively for such elements that produce their corresponding oxides when exposed in the flame, for example : Al, Mo, Si, Ti, W, V. Nevertheless, these estimations may be performed under suitably modified experimental parameters, and
(3) When the solutions of metal salts are made in an aqueous medium the predominant anion present affects the resulting signal to a negotiable extent.
255. What are the important aspects of atomic absorption spectroscopy?
The following three important aspects of atomic absorption spectroscopy:
(i) Analytical Techniques,
(ii) Detection Limit and Sensitivity, and
(iii) Interferences.
256. What are the Analytical Techniques used in Atomic Absorption Spectroscopy?
- Calibration curves
- Standard addition method
257. Which typical interferences are observed in Atomic Absorption Spectroscopy?
(i) Spectral Interferences,
(ii) Chemical Interferences,
(iii) Ionisation Interferences.
258. Applications of Atomic Absorption Spectroscopy.
i. Assay of total zinc in insulin zinc suspension
ii. Assay of palladium in carbenicillin sodium
ii. Cognate assays
Thin-Layer Chromatography (TLC)
259. Explain the theory of Thin-Layer Chromatography (TLC).
The adsorbent used for TLC is a thin, uniform layer (approximate thickness is 0.24 mm) of a dry, finely powdered material applied to an appropriate support, such as a glass plate or an aluminium sheet or a plastic foil.
Thereafter, the mobile phase is permitted to move across the surface of the plate (by capillary action) and the chromatographic phenomenon is depend upon adsorption, partition, or a combination of both, depending on the adsorbent, its treatment, and the nature of the solvents employed.
To carry out the analysis using the chromatographic separation procedure the TLC-plate is placed in a chromatographic chamber, generally made up of glass to enable clear observation of the movement of the mobile phase up the plate, that is pre-saturated with the solvent vapour.
260. Give some examples of Thin-Layer Chromatography (TLC) inert solid supports.
Examples of the inert solid supports are, alumina, silica gel, kieselguhr and cellulose.
261. Why Thin-Layer Chromatography (TLC) is considered more versatile then paper and column chromatography.
(i) Simple equipment: TLC requires simple equipment, such as micro-slides, jars with lid, glass-sprayers, strips of glass sheet, and small chromatank.
(ii) Short development time: The separation is very rapid. The development time is of short duration (around 1 hour) for reasonably good separation on inorganic adsorbent layers.
(iii) Wide choice of stationary phase: TLC may be used for adsorption, partition (including reversed phase) or ion-exchange chromatography,
(iv) Quick recovery of separated constituents: TLC permits the possibility of removal of the adsorbent coating on the plates by scraping with a spatula. In other words, a spot or a zone can be removed quantitatively, and the separated constituent dissolved in an appropriate solvent is estimated either by suitable spectrophotometric or colorimetric analysis.
(v) Separation effects: The separation effects obtained by TLC are more distinctive and superior than those of paper chromatography,
(vi) Easy visualization of separated components: Detection of fluorescence components when exposed to UV light is much easier than on paper by virtue of the fact that inorganic material (i.e., adsorbent) has intrinsic fluorescence,
(vii) Detection Limit: TLC affords extremely sharp delineated spots and offer lower detection limit i.e., one decimal power less than that in paper chromatography,
(viii) Variable thickness of layers: The method employed in TLC may be further extended to preparative separations by using thicker layers and also to meet separations by column chromatography,
(ix) Chemically inert stationary phase: Use of inorganic adsorbents e.g., alumina and silica, in TLC allows the application of corrosive sprays to detect fractionated substances, for instance: carbohydrates by 70% conc. H2SO4,
(x) Trace analysis: TLC method is suitable as micromethod in trace analysis.
262. Explain different techniques for preparation of thin layers on plates.
The most important aspect of preparation of thin layer is that it must be uniform and consistent throughout. Following are different techniques for preparation of thin layers on plates.
(a) Pouring of Layers
(b) Dipping
(c) Spraying
(d) Spreading
(e) TLC-Plates ready-for Use (or Pre-coated Plates)
(a) Pouring of Layers: In order to obtain layers of equal thickness, a measured amount of the suspension or slurry is placed on a given-size plate that is rested on an absolutely labelled surface. The plate is subsequently tipped backward and forward to permit the slurry (or suspension) to spread uniformly on the surface of the plate.
(b) Dipping: In this technique, two plates at a time back-to-back are dipped together in a slurry of the adsorbent in either chloroform or chloroform methanol.
(c) Spraying: In this method use of a small paint-sprayer is used for the distribution of the suspension or slurry onto the surface of the glass-plate.
There are two major disadvantages of using this technique, i.e. (i) Non-uniformity of layers on a single-plate, and (ii) Variation observed from one plate to the other was significant.
(d) Spreading: In this technique the suspension or slurry is put in an ‘applicator’, which is subsequently moved either over the stationary glass-plate or vice-versa i.e., it is held stationary while the glass plate is pulled or pushed through. This technique usually yields uniform thin layers on the glass plates.
263. What are the advantages of ‘ready-for-use’ TLC-plates?
• It can be safely activated at 110-120° C, before, use,
• The properties of the layer minimize spot-diffusion that helps both more strong concentration of spots and more distinctive separations with higher sensitivity,
• It generally accepts more corrosive spray-reagents, for example: conc. sulphuric acid, phosphoric acid, phosphomolybdic acid, perchloric acid on antimony trichloride. Also the sprayed plates could be heated upto 110-120 °C without any darkening,
• The migration rate is slightly enhanced when compared to hand coated plates, and
• The TLC plates may be cut into strips by the aid of a glass cutter applied on the reverse side.
264. What should be considered to choose adequate adsorbent for TLC?
The choice of adsorbent in TLC plays a key role in the separation of components either belonging to natural origin or to purely synthetic origin. Following attributes helps to choose the adsorbent for TLC:
(i) Solubility of the substance e.g., hydrophilic and lipophilic,
(ii) Nature of the compound i.e., whether it is acidic/basic/neutral/amphoteric
(iii) Reactivity of compound with either the solvent or the adsorbent, and
(iv) Chemical reactivity of compounds with the binders.
The adsorbents are of mainly two types, inorganic, and organic adsorbents.
265. What are the inorganic, and organic adsorbents used for TLC?
A. Inorganic adsorbents
(i) Aluminium oxide
(ii) Aluminium Silicate
(iii) Bauxite (aluminium oxide ore)
(iv) Bentonites
(v) Calcium Carbonate
(vi) Calcium Hydroxide
(vii) Calcium Oxalate
(viii) Calcium Silicate
(ix) Calcium Sulphate
(x) Dicalcium Phosphate
(xi) Fuller’s Earth
(xii) Hydoxyl-Apatite
(xiii) Kieselguhr (Diatomaceous Earth)
(xiv) Magnesium Silicate (Magnesol)
(xv) Silica Gel (of pH 6.0)
(xvi) Tri-calcium Phosphate
(xvii) Water-soluble salts
(xviii) Zinc Carbonate
B. Organic Adsorbents
(i) Cellulose and Acetylated Cellulose
(ii) Charcoal and Activated Carbon
(iii) Dextran Gels
(iv) Cellulose Ion-Exchange Powder
(v) Ion-Exchange Resins
(vi) Polyamide
(viii) Sucrose
266. What should be considered to choose a solvent system for TLC?
The choice of solvent or a mixture of solvents used in TLC is depend on two important attributes:
(a) the nature of the constituent to be separated (polar or non-polar)
(b) the nature of the process involved (‘adsorption’ or ‘partition chromatography’)
267. What is meant by activation of adsorbent for TLC?
Activation of the TLC plate means completely eliminating the solvent embedded into the thin layer of coated adsorbent.
It is achieved conveniently first by air-drying the TLC plates for a duration of 30 minutes and then in a hot-air oven maintained at 110 °C for another 30 minutes and subsequently cooling them in a dessicator.
This drying process helps a great extent in rendering the adsorbent layer active. In order to achieve very active layers, silica gel and alumina coated plates may be heated upto 150 °C for a duration of 4 hours and cooling them in a dessicator.
268. Explain the process of purification of silica gel-g layers for TLC?
The iron present as an impurity in silica gel-G affords an appreciable distortion of the ‘chromatogram’. Therefore, it is necessary step to purify the adsorbent.
The ‘iron-free’ layers may be achieved by providing the pre-coated and air-dried plates a preliminary development with a mixture of methanol and concentrated hydrochloric acid (9 : 1).
By this process the entire iron gets migrated with the solvent front to the upper boundary of the TLC plate. Therefore, the purified plates are again dried and activated at 110°C.
The cleaning process usually washes out the CaSO4 originally present as binder. Hence, the silica gel thus obtained by purification may be reused to prepare TLC-plates with other appropriate binders like gypsum, starch etc.
269. What is Equilibration of the Chamber in TLC and why it is important?
The equilibration of the chamber or chamber-saturation is a vital factor to obtain reproducible Rf values.
Equilibration of the chamber is achieved by allowing the solvent system to remain in the chamber for at least 1 to 2 hours so that the vapours of the solvent(s) would pre-saturate the latter adequately.
This is done to obtain distinct separation of constituents, uniform solvent from and prevent evaporation of the solvent on TLC-plates.
270. How to protect TLC experiment from Oxidation?
Both temperature and light augments oxidation and, hence, the following experimental Conditions can b maintain to obtain the best development of thin-layers,
Temperature: 18-23°C, and Light : Diffused daylight both natural and artificial,
Note: Direct sunlight (UV) or drought may give rise to ‘oblique formation’ of the solvent front.
271. What is the meaning of visualization in the TLC experiment?
As a result of both intensive as well as extensive research a number of organic and inorganic substances have been identified that positively demonstrate an ‘improved visualization’. Such substances are termed collectively as ‘fluorescent indicators’.
272. Examples of fluorescent indicators in the TLC experiment to aid visualization?
Following are examples of fluorescent indicators in the TLC experiment to aid visualization
- Barium diphenylamine sulphonate
- 2,7-dichlorofluorescein
- Fluorescein (0.2% w/v in Ethanol)
- Morin (0.1% w/v in Ethanol)
- Sodium fluorescinate (0.4% w/v in water)
- Rhodamine B
- Zinc Silicate
- Calcium silicate
- Methylumbelliferone (or 7-hydroxy-4-methyl coumarin)
273. Explain various special techniques of TLC.
(a) Horizontal TLC
(2) Continuous TLC
(3) Preparative TLC
(4) Multiple Dimensional TLC
(5) Two-Dimensional Chromatography
(6) Centrifugal Chromatography
(7) Wedged-Tip Chromatography
274. Explain Horizontal TLC technique.
In this technique, the horizontal development of loose-layer TLC plates were made by using a shallow dish having a ground glass cover. The TLC plate was carefully rested on a T-shaped glass piece and the starting end was pressed duly against a filter paper held by another glass strip, which allowed the solvent to move to the thin-layer-film from the bottom of the dish by capillary action.
275. Explain Continuous TLC technique.
Continuous TLC technique is good for the separation of such components having small as well as very close Rf values. Following are the technique:
(a) Rectangular horizontal plates where the solvent is allowed to move over them and subsequently evaporated after it has almost reached the end of the run, and
(b) Triangular glass-plates-where the mixture to be separated is spotted near the apex on a thinlayer and two different solvent mixtures are fed from two sides to the thin-layer and fractions
subsequently collected at the base.
276. Explain Preparative TLC technique.
TLC may be skillfully extended to cater for extremely useful method for preparative separations. To maintain uniformity, as a rule, plates of 20 cm height and 20-100 cm length with layers between 0.5 and 0.2 mm thickness are normally employed. It essentially has three cardinal features, namely:
(a) Component mixtures is always obtained either in streaks or bands,
(b) Separation is invariably accomplished by multiple development, and
(c) Localization of separated components is only done under UV-light.
277. Explain Multiple Dimensional TLC technique.
Multiple Dimensional TLC technique can be regarded as a variant of multiple development chromatography.
278. Explain Two-Dimensional Chromatography TLC technique.
It is also termed as two-dimensional planar chromatography. Here, the sample is spotted in one corner of a square TLC plate (size : 20 cm × 20 cm). The development is first carried out in the ascending direction using solvent-1. The solvent is then eliminated by evaporation and the plate is rotated through 90°, following which ascending with the second solvent is accomplished.
After Removal of the solvent the spots of separated constituents are located by spraying with specific reagents.
279. Explain Centrifugal Chromatography TLC technique.
It essentially makes use of the ‘centrifugal force’ so as to accelerate the flow of solvent through the thin-layer of the chromatogram. The sample mixture is applied 2.5 cm from the centre hole and the solvent system is set to allow a constant flow, with the centrifuge rotating at 500-700 RPM. In this manner, the usual developing time of 35 minutes is drastically reduced to mere 10 minutes by acceleration.
280. Explain Wedged-Tip Chromatography technique of TLC.
This technique exhibit the following two plus points, namely :
(a) Improved separation, and
(b) Constituents forced to assume an almost band-like path.
For TLC-plate with wedged-tip, following steps are to be adopted sequentially:
(i) Draw dividing lines 0.5 to 1.0 mm broad on the surface of the layer with a narrow-metal spatula,
(ii) Pentagons are facilitated by the help of a stencil made of transparent plastic material, and
(iii) Sample mixture are applied to the narrow portion of the wedge to get the best results.
281. Explain the process of detection of components in TLC.
After development of TLC plates, the next important step is to detect the separated components so as to determine their respective Rf values. It can be achieved using following techniques:
(i) Coloured Substances: e.g., Xanthophylls, Chlorophylls, Carotenes, etc., may be located visually.
(ii) Colourless Substances: e.g., alkaloids, steroids, amino acids and the like may be detected under short-wave UV-light or a long-wave UV-light. These substances may also be detected as brown/dark brown spots when exposed to I2-vapours in a closed dessicator.
(iii) Specific Detecting Reagents: A few specific detecting reagents are normally used for a particular class of compounds e.g., Aniline-phthalate reagent : for carbohydrates; Ninhydrin reagent: for amino-acids, and Dragendorff’s reagent: for alkaloids
(iv) Chromic acid/conc. H2SO4: These corrosive reagents usually char the organic material on TLC plates and may be seen as dark brown spots.
282. Explain the process of evaluation of the chromatogram in TLC.
After completing the detection procedure the various separated solutes on the TLC plate are marked with the help of a sharp needle (e.g., pithing needle); subsequently, their evaluation could be carried out either qualitatively or quantitatively.
A. Qualitative Evaluation:
The Rf value (Retention Factor) various separated solutes is determined accurately. The Rf value represents the differences in rate of movement of the components duly caused by their various partition coefficients i.e., their different solubility in the mobile and stationary phases. In order words, the Rf value (relate to front) is-‘the ratio between the distance starting point-centre of spot and distance starting point-solvent front’, thus it may be expressed as :
Rf = Distance of centre of spot from starting point/ Distance of solvent front from starting point.
Characteristics of Rf value:
(i) Due to the always longer path of the solvent front, the Rf value is invariably lesser than 1.
(ii) Rf value is always constant for each component only under identical experimental parameters,
(iii) Rf value depends upon a number of governing factors, such as: quality of the layer material; activation grade of the layer ; thickness of layer; quality of solvent; equilibration of chamber; chromatographic technique employed (e.g., ascending, descending); presence of impurities; and conc. of simple applied; and
(iv) All possible anomalies in (iii) above may be eliminated by performing a co-chromatogram of a standard substance along with that of a sample. Thus, the distance traversed by a substance is compared with that of the standard (or reference). This ‘new’ relation is usually designated as Rst value.
Therefore, in short, it is expressed as follows :
Rst = Rf of the substance/ Rf of the standard
Unlike the Rf value, the Rst value may be more than 1.00 because here the substance under investigation (i.e., sample) usually travels further than the standard. In TLC, the qualitative evaluation is solely based on the determination of Rf values of unknown spots vis-a-vis Rf values of standard substances preferably on the same TLC plate so as to avoid any possible error whatsoever.
B. Quantitative Analysis:
The quantitative analysis of chromatographically separated constituents may be carried out with high degree of accuracy and precision in two manners, namely :
(i) Direct Method : i.e., the quantitative determinations is performed directly on the adsorbent layer, and
(ii) Indirect Method : i.e., the separated constituents are quantitatively removed from, the adsorbent and subsequently estimated after elution.
(a) Direct Methods:
The various methods under this category are, namely :
(i) Measurement of Spot-areas : This method is solely based on a mathematical relationship existing between the prevailing spot area and the amount of component present. It is not quite accurate due to high random errors.
(ii) Densitometry : The intensity of the colour of a component is measured on the chromatogram using a densitometer.
(iii) Spectrophotometry : Characterization of the separated spots by reading the absorption or fluorescence curves directly from TLC plates is carried out with the help of Chromatogram Spectrophotometer devised by Zeiss, Stahl and Jork.
Besides, IR-spectroscopy, reflectance spectroscopy, spark chamber method etc., may also be employed for the direct evaluation of chromatograms.
(b) Indirect Methods:
These methods are based on elution techniques, followed by micro-analysis of the resultant eluate by adopting one or more of the undermentioned known methods, namely: Colorimetry; Fluorimetry ; Radiometry; Flame-photometry; UV-Spectrophotometry; Gravimetry; Polarography; Vapourphase Chromatography ;
283. Explain the applications of TLC in pharmaceutical analysis.
The technique of thin-layer chromatography (TLC) has been used extensively in the domain of pharmaceutical analysis for a variety of specific and useful applications, for example :
(i) To identify the presence of undesirable specific organic compounds present as impurities in a number of pharmaceutical substances, namely : morphine in apomorphine hydrochloride ; hydrazine in carbidopa ; 3-aminopropanol in dexampanthenol ; etc.,
(ii) Related substances present in official drugs, namely : related substances present in a wide number of potent pharmaceutical substances e.g., aminophylline ; baclofen ; chloramphenicol ; carbamazepine etc.,
(iii) Foreign alkaloids present in alkaloidal drugs, for instance : atropine sulphate ; codeine;
(iv) Foreign steroids present in steroidal drugs, for example : betamethasone valerate;
(v) Ninhydrin positive substances in official amino acids e.g., glutamic acid; leucine;
HPLC Interview Questions and troubleshooting
Click Here to refer Most useful 80+ HPLC Interview Questions and troubleshooting
Refer Complete guide on High Performance Liquid Chromatography (HPLC)
For more interview questions and answers Click Here
Reference – Pharmaceutical Drug Analysis by A-s-h-u-t-o-s-h K-a-r, Wikipedia and other relevant textbooks