
Top 10 important Interview Questions and Answers on Pharmaceutical Validation

This page covers most of the interview questions and answers asked during a technical interview round of quality assurance and validation professionals.
You will find interview questions and answers on Terminologies associated with process validation, Stages of Process Validation, approach to process validation, Stages of process validation, typical steps for QbD, control strategy of process validation, FDA guidance, EMA guidance, WHO guidance on hold time studies of the products, different guidelines/ regulations describing requirement of cleaning validation, and different guidelines/ regulations describing requirement of equipment qualification.
The interview questions cover questions from basic to advance level of technical aspects. These interview questions and answers will help to crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
The topics covered here are the sterilization process, aseptic processing, media fill, area classification, and associated topics. In addition, the interview questions and answers cover various equipment used for the manufacturing process of sterile formulations.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning, and best of luck with your interview! Happy Learning.
1. Terminologies associated with process validation.
i. Attribute
An attribute is a physical, chemical, or microbiological property or characteristic of an input or output material.
Reference: ICH Quality Guideline Q5E
ii. Critical Quality Attribute (CQA)
A CQA is a physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
Reference: ICH Quality Guideline Q8 (R2)
iii. Quality Attribute
A Quality Attribute is a molecular or product characteristic that is selected for its ability to indicate the quality of the product. Collectively, the quality attributes define identity, purity, potency and stability of the product, and safety with respect to adventitious agents. Specifications measure a selected subset of the quality attributes.
Reference: ICH Quality Guideline Q6B
iv. Control Strategy
A control strategy is a planned set of controls, derived from current product and process understanding that assures process performance and product quality (ICH Q10).
Every drug substance manufacturing process, whether developed through a traditional or an enhanced approach (or some combination thereof), has an associated control strategy.
A control strategy can include, but is not limited to, the following:
Controls on material attributes (including raw materials, starting materials, intermediates, reagents, primary packaging materials for the drug substance, etc.);
Controls implicit in the design of the manufacturing process (e.g., sequence of purification steps (Biotechnological/Biological Products), or order of addition of reagents (Chemical Products));
In-process controls (including in-process tests and process parameters); Controls on drug substance (e.g., release testing).
Reference: ICH guideline Q11
v. Continued Process Verification (CPV)
The CPV is the Stage 3 of Process Validation. The goal of this stage is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture.
This is science and risk-based approach for collection and evaluation of information and data about the performance of the process, which will allow detecting undesired process variability. Evaluating the performance of the process identifies problems and determines whether action must be taken to correct, anticipate, and prevent problems so that the process remains in control (§ 211.180(e)).
vi. Critical process parameter (CPP):
A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality.
Reference: ICH guideline Q8
vii. Critical Material Attribute (CMA)
Critical material attribute (CMA) are defined as A material whose variability (physical, chemical, biological or microbiological property or characteristic of an input material) has an impact a critical quality attribute and therefore it should be monitored or controlled to ensure desired drug product quality.
Reference: http://www.rsc.org/
viii. Design Space
The design space is the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality.
Working within the design space is not considered a change. Movement out of the design space is considered to be a change, and would normally initiate a regulatory post-approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.
Reference: ICH guideline Q8 (R2)
ix. Formal Experimental Design (synonym: design of experiments)
A Formal Experimental Design is a structured, organized method for determining the relationship between factors affecting a process and the output of that process.
Reference: ICH guideline Q8 (R2)
x. Lifecycle
Lifecycle includes all phases in the life of a product, from the initial development through marketing until the product’s discontinuation.
Reference: ICH guideline Q8 (R2)
xi. Normal Operating Range (NOR)
The NOR is a defined range, within (or equal to) the Proven Acceptable Range, specified in the manufacturing instructions as the target and range at which a process parameter is controlled, while producing unit operation material or final product meeting release criteria and CQAs.
Reference: Process Robustness – A PQRI White Paper, Pharma. Engin. 2006.
xii. Key Process Parameter (KPP; synonym: key operational parameter)
This is an input process parameter that should be carefully controlled within a narrow range and is essential for process performance. A key process parameter does not affect product quality attributes. If the acceptable range is exceeded, it may affect the process (e.g., yield, duration) but not product quality.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xiii. Non-Key Process Parameter (Non-KPP; synonym: non-key operational parameter)
This is an input parameter that has been demonstrated to be easily controlled or has a wide acceptable limit. Non-key operational parameters may have an impact on quality or process performance if acceptable limits are exceeded.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xiv. Process Parameter (synonym: operational parameter)
This is an input variable or condition of the manufacturing process that can be directly controlled in the process. Typically, these parameters are physical or chemical (e.g., temperature, process time, column flow rate, column wash volume, reagent concentration, or buffer pH).
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xv.Platform Manufacturing
This means the development of a production strategy for a new drug starting from manufacturing processes similar to those used to manufacture other drugs of the same type (the production for which there already exists considerable experience).
Reference: ICH guideline Q11
xvi.Process Analytical Technology (PAT)
A PAT is a system for designing, analyzing, and controlling manufacturing through timely measurements (i.e., during processing) of critical quality and performance attributes of raw and in-process materials and processes with the goal of ensuring final product quality.
Reference: ICH guideline Q8 (R2)
xvii.Process Performance Qualification (PPQ)
This is the second element of Process Qualification. It includes a combination of the actual facility, utilities, equipment, and trained personnel with the commercial manufacturing process, control procedures, and components to produce commercial batches. A successful PPQ will confirm the process design and demonstrate that the commercial manufacturing process performs as expected. Batches prepared are also called ‘Conformance batches’ or ‘PPQ batches’.
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
xviii.Process Qualification
This qualification confirms that the manufacturing process, as designed, is capable of reproducible commercial manufacturing..
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
It consists of following 2 important elements:
(i) Qualification of Facility, Equipment, and Utilities
(ii) Process Performance Qualification
xix. Process Robustness
Ability of a process to tolerate variability of materials and changes of the process and equipment without negative impact on quality is known as process robustness.
Reference: ICH guideline Q8 (R2)
xx. Process Validation
As per US FDA
Such validation is the collection and evaluation of data from the process design stage to commercial production, which establishes with scientific evidence that a process is capable of consistently delivering quality products.
Reference: Guidance for Industry: Process Validation: General Principles and Practices; U.S. Food and Drug Administration: 2011.
As per EMA
Such validation comprises documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes.
Reference: Draft Guideline on Process Validation, MA/CHMP/CVMP/QWP/70278/2012-Rev1; European Medicines Agency: 2012.
xxi. Prospective approach to PPQ
This indicates an approach wherein the Process Performance Qualification batches, manufactured using a qualification protocol, are released for distribution only after complete execution of the Process Performance Qualification Study
Reference: ISPE Guidance
xxii. PPQ re-verification
This indicates the repeating of a part of or a complete PPQ study in the event of changes in the process, equipment, etc. or as a recommendation of the CPV process to verify whether a process continues in a validated state of control and/or to verify that the changes do not adversely impact process characteristics and product quality or the validated state of control of the process
Reference: ISPE Guidance
xxiii. Product Lifecycle
All phases of product stats from the initial development through marketing until the product discontinuation.
xxiv. Process Validation Master Plan (synonym: validation master plan)
This is a document that defines the process validation scope and rationale and that contains the list of process validation studies to be performed.
Reference: Technical Report No. 42: Process Validation of Protein Manufacturing; Parenteral Drug Association: 2005.
xxv. Quality
This indicates the suitability of either a drug substance or drug product for its intended use. This term includes such attributes as the identity, strength and purity.
Reference: ICH guideline Q6A
xxvi. Quality by Design (QbD)
This means a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
Reference: ICH guideline Q8 (R2)
xxvii. Quality Target Product Profile (QTPP)
QTPP is a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product.
Reference: ICH guideline Q8 (R2)
xxviii. Verification
Verification is a systematic approach to verify that manufacturing systems, acting alone or in combination, are fit for intended use, have been properly installed, and are operating correctly. This is an umbrella term that encompasses types of approaches to ensure that the systems are fit for the designed purpose. Other terms used are qualification, commissioning and qualification, system validation, etc.
Reference: ASTM E2500-07. Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment; American Society for Testing and Materials: 2007.
xxix. Worst Case
A set of conditions encompassing upper and lower processing limits and circumstances, including those within standard operating procedures, that pose the greatest chance of process or product failure (when compared to ideal conditions). Such conditions do not necessarily induce product or process failure.
Reference: EudraLex: The Rules Governing Medicinal Products in the European Union: Volume 4 Good Manufacturing Practice Medicinal Products for Human and Veterinary Use, Annex. 15.
Qualification and Validation; European Commission: 2014.
2. What are the Stages of Process Validation?
Stage 1 – Process Design
Stage 2 – Process Qualification
Stage 3 – Continued Process Verification
3. What is an integrated team approach to process validation?
Integrated team approach to process validation that includes expertise from a variety of disciplines (e.g., process engineering, industrial pharmacy, analytical chemistry, microbiology, statistics, manufacturing, and quality assurance).
4. Explain “Process Design”, Stage 1 of process validation.
Objective: To design a process that can consistently deliver a commercial product meeting quality attributes.
a. Building and Capturing Process Knowledge and Understanding from
• Previous experience with similar processes
• Product and process understanding (from clinical and pre-clinical activities)
• Analytical characterization
• Published literature
• Engineering studies/ batches
• Clinical manufacturing
• Process development and characterization studies
• Product development activities
• Design of Experiment (DOE) studies to develop process knowledge and relationships between the variable inputs (e.g., component characteristics 13 or process parameters) and the resulting outputs (e.g., in-process material, intermediates, or the final product)
• Computer-based or virtual simulations of certain unit operations or dynamics can provide process understanding and help avoid problems at commercial scale
• Documentation of process understanding, activities and studies
b. Establishing a Strategy for Process Control
An appropriate control strategy is based on knowledge and experience gained in Stage 1 that will help to control the manufacturing process.
Strategy for Process Control includes the following elements:
• Raw material controls
• In-process and release specifications
• In-process controls
• Performance parameters
• Process parameter set points and ranges
• Process monitoring (data review, sampling, testing)
• Processing and hold times
• Process Analytical Technology (PAT)
5. In which scenarios process control through operational limits and in-process monitoring is essential?
In case of following two possible scenarios, process to be controlled using operational limits and in-process monitoring:
a. When the product attribute is not readily measurable due to limitations of sampling or detectability (e.g., viral clearance or microbial contamination) or
b. When intermediates and products cannot be highly characterized and well-defined quality attributes cannot be identified.
6. Explain “Process Qualification”, Stage 2 of process validation.
Objective: In this stage, the process design is evaluated to determine if it is capable of reproducible commercial manufacture.
This stage has two elements:
i. Design of the facility and qualification of the equipment and utilities
ii. Process performance qualification (PPQ).
a. PPQ Protocol
b. PPQ Protocol Execution and Report
CGMP-compliant procedures must be followed. Successful completion of Stage 2 is necessary before commercial distribution. Products manufactured during this stage, if acceptable, can be released for distribution.
7. Explain “Continued Process Verification”, Stage 3 of process validation.
Objective: The goal of the third validation stage is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. A system or systems for detecting unplanned departures from the process as designed is essential to accomplish this goal.
8. Explain typical steps for QbD steps.
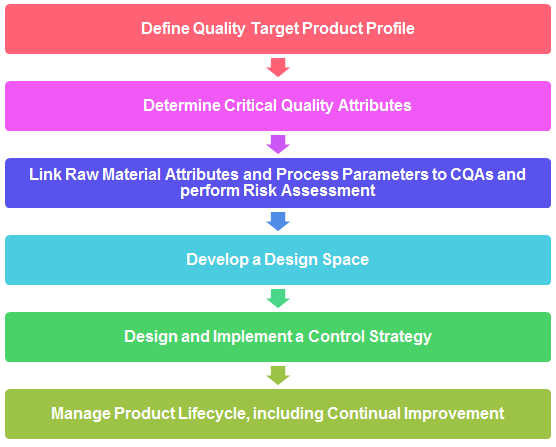
9. What is Quality Target Product Profile (QTPP)? Explain with examples.
The QTPP is defined by capturing all relevant quality requirements of the drug product to be developed.
It consists of following:
i. Dosage form and strength, route of administration, delivery systems, container and closure system etc.
ii. Drug substance quality attributes required for intended drug product, i.e. physical, chemical, and biological properties
iii. Drug product quality attributes required for dosage form, i.e. physical, chemical and microbiological attributes of drug products.
iv. Bioavailability attributes, i.e. dissolution requirement or other relevant characteristics. v. Excipient quality attributes, input material compatibility, stability, pharmacology, etc.
10. What is a control strategy? What shall be included while defining control strategy?
• Raw material controls
• In-process and release specifications
• In-process controls
• Performance parameters
• Process parameter set points and ranges
• Process monitoring (data review, sampling, testing)
• Processing and hold times
• Process Analytical Technology (PAT)
11. Which FDA guidance or Code of Federal Regulation suggests having hold time studies of the products?
Establishing production time limits is an example of a control to prevent growth of objectionable microorganisms. Per 21 CFR 211.111, time limits for the completion of each phase of production, when appropriate, must be established and followed. For example, if a firm finds it necessary to hold a bulk topical or liquid product for several months until it is filled, the firm might establish a holding time limit to help prevent objectionable microbial buildup. Validation and control over microbial content of purified water systems used in certain topical products are also examples of such procedures (see FDA guidance, referenced below).
References:
21 CFR 211.113: Control of microbiological contamination
21 CFR 211.165: Testing and release for distribution
21 CFR 211.111: Time limitations on production
FDA Guidance for Industry, 2011, Process Validation: General Principles and Practices
12. Which EMA guidance suggests having hold time studies of the products?
4 July 2017, EMA/CHMP/QWP/245074/2015, Committee for Human Medicinal Products (CHMP), Guideline on manufacture of the finished dosage form
As per guideline
“Depending on the nature of the process and the product (e.g. sterile products), manufacturing durations of critical steps and hold times should be stated and justified”.
“Where relevant, the maximum holding times of the bulk product or, alternatively, the maximum batch manufacturing time from start of product manufacture to completion of packaging into the final primary container for marketing should be stated, appropriately justified and supported by data in relevant parts of the dossier (e.g. challenging the maximum hold time in process validation studies or providing dedicated stability studies for the bulk storage)”.
13. Which WHO guidance suggests having hold time studies of the products?
WHO Technical Report Series No. 992, 2015, Annex 4, General guidance on hold-time studies.
As per guidance
“Normally, intermediate and bulk products should not be stored beyond the established hold time. The choice of maximum holding period should be supported by relevant data. Studies may extend beyond the chosen maximum but it is not necessary to extend testing to determine the extreme limits at which failure occurs”.
14. What are the different guidelines/ regulations describing requirement of cleaning validation?
A. U.S. Food and Drug Administration (U.S. FDA)
PART 211 — CURRENT GOOD MANUFACTURING PRACTICE FOR FINISHED PHARMACEUTICALS – Subpart D – Equipment
Source: Sec. 211.67 Equipment cleaning and maintenance.
Questions and Answers on Current Good Manufacturing Practices – Equipment
GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES
Source: https://www.fda.gov/validation-cleaning-processes-793
B. European Medicines Agency (EMA)
20 November 2014, EMA/CHMP/ CVMP/ SWP/169430/2012, Committee for Medicinal Products for Human Use (CHMP), Committee for Medicinal Products for Veterinary Use (CVMP), Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities
19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018 Committee for Medicinal Products for Veterinary Use (CVMP) Committee for Medicinal Products for Human Use (CHMP) Questions and answers on implementation of risk-based prevention of cross-contamination in production and ‘Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities’ (EMA/CHMP/CVMP/SWP/169430/2012)
C. World Health Organization (WHO)
QAS20 849 Points to consider on the different approaches –including HBEL – to establish carryover limits in cleaning validation for identification of contamination risks when manufacturing in shared facilities
WHO good manufacturing practices for active pharmaceutical ingredients
Source: https://www.who.int/medicines/areas/quality_safety/quality_assurance/GMPActivePharmaceuticalIngredientsTRS957Annex2.pdf
Quality assurance of pharmaceuticals: A compendium of guidelines and related materials
Source: https://www.who.int/medicines/areas/quality_safety/quality_assurance/QualityAssurancePharmVol2.pdf
Points to consider when including6 Health-Based Exposure Limits (HBELs) in cleaning validation
D. Pharmaceutical Inspection Co-operation Scheme (PIC/S)
Cross-contamination in shared facilities (PI-043-1)
Source: https://picscheme.org/docview/2270
Guideline on Setting HBEL for use in risk identification in the manufacture of different medicinal products in shared facilities (PI 046-1)
Source: https://picscheme.org/docview/2467
Guide to GMP for medicinal products Part 1 (PE 009-14 (Part I))
Source: https://picscheme.org/docview/4205
Inspection of Health Based Exposure Limit (HBEL) Assessment and use in Quality Risk Management (PI 052-1)
Source: https://picscheme.org/docview/1947
E. Therapeutic Goods Administration (TGA)
The TGA is adopting version PE009-13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products (PIC/S Guide to GMP)
Source: https://www.tga.gov.au/sites/default/files/transition-new-gmp-requirements-medicinal-products.pdf
TGA interpretation and expectations for demonstrating compliance
Source: https://www.tga.gov.au/resource/pe009-pics-guide-gmp-medicinal-products
A presentation on Cleaning Validation by TGA
Source: https://www.tga.gov.au/sites/default/files/presentation-cleaning-validation.pdf
F. Health Canada
Cleaning validation guide (GUI-0028)
G. Active Pharmaceutical Ingredients Committee (APIC)
Guidance on aspects of Cleaning Validation in Active Pharmaceutical Ingredient Plants APIC Cleaning Validation 2016
Source: https://apic.cefic.org/pub/APICCleaningValidationGuide-updateSeptember2016-final.pdf
Guidance on aspects of Cleaning Validation in Active Pharmaceutical Ingredient Plants APIC Cleaning Validation 2021
Source: https://apic.cefic.org/publications/APIC_Cleaning-validation-guide_2021.pdf
H. Parenteral Drug Association (PDA)
PDA Technical Report 29: Points to Consider for Cleaning Validation
PDA Technical Report 49: Points to Consider for Biotechnology Cleaning Validation
I. International Society For Pharmaceutical Engineering (ISPE)
Baseline Guide Vol 7: Risk-Based Manufacture of Pharma Products 2nd Edition ISPE Risk-MaPP
Cleaning Validation Lifecycle – Applications, Methods, and Controls ISPE Cleaning Validation Guideline
J. American Society For Testing and Materials (ASTM)
ASTM E3106 – 18e1 (Standard Guide for Science-Based and Risk-Based Cleaning Process Development and Validation)
Standard Guide for Derivation of Health-Based Exposure Limits (HBELs) ASTM E3219
15. What are the different guidelines/ regulations describing requirement of equipment qualification?
a. EU GMP Annex 15 “Qualification and Validation”, Eudralex 2015
b. PIC/S GMP for Medicinal Products Annex 15 “Qualification and Validation” PE009-14 (Annexes) 2018
c. PE 009-15 (Annexes) ANNEX 15 – Qualification and validation
d. PI 006-3 – Validation Master Plan installation and operational qualification non-sterile process validation cleaning validation
e. Subpart C – Building and Facilities Section 211.42 – Design and Construction Features
f. USP 37: “Analytical Instrument Qualification”, (section 1058), The United States Pharmacopeial Convention, 2016
g. ISPE Baseline Guide Volume 5: “Commissioning & Qualification”, Second Edition, 2019
h. ASTM E2500: “Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment”, ASTM 2020
i. ISPE Good Practice Guide: Applied Risk Management for Commissioning and Qualification, ISPE 2011
j. PDA Technical Report 54-5: “Quality Risk Management for the Design, Qualification, and Operation of Manufacturing Systems”, PDA 2017
k. EU GMP Annex 11 “Computerised Systems”, Eudralex 2011
l. US FDA Guidance for Industry: “Process Validation: General Principles and Practices, 2011
m. ISPE GAMP® 5 Guide: “A Risk-Based Approach to Compliant GxP Computerized Systems”, ISPE 2008
n. ISPE Guide: “Science and Risk-Based Approach for the Delivery of Facilities, Systems, and Equipment”, ISPE 2011
o. FDA US CFR 21 part 11 Electronic Records; Electronic Signatures 2003
p. Health Canada: Guide to validation – drugs and supporting activities (GUI-0029)
q. WHO Technical Report Series, No. 937, Annex 4: Supplementary guidelines on good manufacturing practices: validation