
Most useful 80+ HPLC Interview Questions and troubleshooting

HPLC interview questions are the most commonly asked topic during the quality control laboratory interview for the pharmaceutical industry. HPLC is the pharmaceutical industry’s most widely used technique of analysis; therefore, most of the interview questions and answers are around HPLC technique and HPLC troubleshooting.
You will find interview questions and answers on components of an HPLC system, isocratic and a gradient HPLC system, types of gradient mixing, function of the HPLC column, types of sample injector, function of the HPLC column oven, commonly used detector in HPLC systems, types of detectors used in HPLC systems, typical HPLC startup procedure, procedure to flush the new reversed-phase column, checkpoints before starting an HPLC analysis, relative retention time (RRT) in liquid chromatography, ‘fronting’ or ‘leading’ in liquid chromatography, peak symmetry, signal-to-noise ratio, procedures for mobile phase degassing, advantages of acetonitrile in the mobile phase, advantages of methanol in the mobile phase, reasons for variable Retention Times (RT) in HPLC analysis, and many other relevant and valuable HPLC interview questions and answers.
The interview questions cover questions from basic to advanced levels of technical aspects. These HPLC interview questions and answers will help crack an interview, enhance your knowledge, and also be helpful for the interviewer who is involved in the recruitment process.
You will find it much more enjoyable while going through these interview questions and answers. So enjoy learning and best of luck with your interview! Happy Learning.
Following are most useful HPLC Interview Questions and answers as well as HPLC troubleshooting guide
1. What are the essential components of an HPLC system?
HPLC instrument consists:
- An eluent delivery system (= pump)
- An injector, a column
- A detector and a data evaluation system
2. What is the difference between an isocratic and a gradient system?
It can be distinguished easily. If there is only one inlet tube for the eluent, it is an isocratic instrument, and, there are two or more are present, its a gradient system.
3. What is the gradient HPLC system?
In a gradient system, two or more solvents are continuously mixed during the separation.
4. What are the types of gradient mixing?
(a) When mixing of solvent happens before the pump by a proportional valve, it is a low pressure gradient.
In a low pressure gradient system, the mixing happens in the normal pressure or low pressure side of the device before the pump.
(b) When the mixing happens after the pump on the high pressure side, mixing takes place in a mixing chamber where the solvents of both pumps meet. Such an instrument has a high pressure gradient.
5. What are the types of sample injector?
(a) Sample introduction with a hand injector or a manual valve
(b) Sample introduction with an autosampler
6. What is the function of the HPLC column?
The HPLC column is the heart of the system. The function of the HPLC column is to separate the compound with various separation mechanisms.
7. What is the function of the HPLC column oven?
Function of the column oven is to maintain a constant temperature of the column and support to get reproducible results.
8. What is the most commonly used detector in HPLC systems?
UV detector and diode array detector (also called as PDA – Photodiode Array).
9. What are the various types of detectors used in HPLC systems?
(i) UV-Visible HPLC Detector
(ii) PDA Detectors (Diode Array Detector or Photo Array Diode Detector) HPLC detector
(iii) Refractive-Index HPLC Detector
(iv) Evaporative Light Scattering Detector (ELSD) HPLC Detector:
(v) Multi-Angle Light Scattering Detector (MALS) for HPLC:
(vi) HPLC-Mass Spectrometer (HPLC-MS) Detector
(vii) HPLC Conductivity Detector:
(viii) HPLC-Fluorescence Detector:
(ix) Chemiluminescence HPLC Detector:
(x) HPLC Optical Rotation Detector or Chiral Detector
(xi) Electrochemical (Amperometric) Detector for HPLC
(xii) HPLC Photoconductivity Detectors
(xiii) HPLC Infrared (IR) Detectors
(xiv) Laser-Induced Fluorescence Detector
(xv) Radioactivity Detector
(xvi) HPLC-NMR Detector
10. What is used to transfer the mobile phase from one module to another?
The mobile phase is transferred from one module to the another module through capillaries made up of stainless steel or PEEK (polyetheretherketone).
11. What is the internal diameter of the HPLC capillaries?
The typical internal diameter of the HPLC capillaries between pump and injector is 0.5-1 mm. The typical internal diameter of the HPLC capillaries after the outlet of the injector is less than 0.2 mm.
12. What is the internal diameter of the HPLC capillaries when back pressure is required to achieve?
Detectors such as fluorescence detectors need some back pressure for adequate operation which can be achieved with a 0.1-0.2 mm capillary internal diameter.
13. What are restrictor capillaries, sometimes also simply called or restrictors?
A restriction capillary or restrictors are the capillaries with very narrow internal diameter, that restrict the mobile phase flow. It has the capability to closely mimic the normal operating conditions by generating back-pressure of 1k to 2k psi.
14. What is the reason for using interconnection pieces of HPLC from the same manufacturer?
Interconnection pieces of HPLC such as ferrules and fittings should be used from the same manufacturer, because while using different make, they may have different dimensions and internal diameters leading to a small dead volume. This can cause abnormality during separation..
15. What is the typical HPLC startup procedure?
a. Flush system without column at a flow rate of 1 ml/min with a 50/ 50 mixture isopropanol/ water for about 10 min.
b. Inject the mobile phase a few times in order to ensure that the old eluent or impurities are removed from the sample injection system.
16. What precautions should be considered before the first sample run in the HPLC?
i. Once the mobile phase is prepared and run in the system, leave the HPLC system for a little time to equilibrate to flush impurities or dirt if any out of the column.
ii. Dissolve samples adequately as per method of analysis.
iii. No particles should remain in the sample, use membrane filtration as per method.
iv. Ensure that your dissolved sample should not precipitate in the mobile phase.
v. Inject a standard and take a look at the chromatogram.
vi. Ensure that the baseline is stable with no drift and peaks are symmetrical.
vii. Ensure that the chromatogram after the second injection is identical to the first one.
17. How to flush the new reversed-phase column?
While installing a new reversed-phase column, flush it with acetonitrile or methanol before use it for the first run.
18. How to ensure that system is free of buffers?
Flush the system first with an isopropanol/ water mixture and then with methanol.
19. What is the procedure to temporarily stop the HPLC (When you are aware that you will run the HPLC on the next day or after a pause)?
When you know that you will run the HPLC (same setup) after some time, shut down all components of the instrument except the pump. Keep the pump running at a lower flow rate about 0.1-0.3 ml/min.
Ensure that sufficient mobile phase is available. Do not run HPLC dry. While resuming the HPLC again, adjust the flow as per the method.
20. What is the procedure to stop the HPLC?
When you want to stop HPLC for a longer duration, flush the system using water to remove the buffer out from the system. Followed by flushing the HPLC using 20-30 ml methanol or acetonitrile.
Store the column using acetonitrile or methanol. Close the column with end fittings to prevent drying of the stationary phase.
21. What is the preferred solvent to store the column if the column need not be used for a longer time? Why?
Acetonitrile is a preferred solvent as it is stable for a longer period of time compared to methanol. Methanol has the property of hydrolysis.
22 What are the checkpoints before starting an HPLC analysis?
1. Ensure no electrical connection is loose.
2. Verify the capillaries for absence of leakages.
3. Check the tubing and solvent container for absence of air bubbles. If any air bubbles are observed, remove air from tubes by sucking solvent with the help of a syringe by opening the purge valve. Perform the purging and priming of the system.
4. Ensure that the solvent container is closed to prevent objects falling into it as well as evaporation of solvents.
5. Check the system flow by switching the pump and ensure that solvent is coming from mobile phase container/ solvent containers to the waste container.
6. Probable reasons of absence of flow are:
a. Air in pump
b. Verify the leakage by touching the seals. It should not be wet.
c. Crystals of buffered mobile phases.
d. Malfunctioning can be verified through unusual pump noise
7. Ensure that mobile phase is prepared using HPLC grade solvents
8. Ensure that the mobile phase is prepared by counting required number of injections, run time and flow rate.
9. Ensure that buffered mobile phases are filtered using a membrane filter, degas with helium or with degasser.
10. While using a manual injector ensure that the container is kept under the overflow.
11. Maintain the injection needles clean to avoid contamination when using manual injector.
12. Clean injector needle using suitable solvent if needed.
13. Use a washing solution while using an autosampler.
14. Check the UV detectors lamp energy when using the UV detector.
15. For RP-HPLC, use 10-20% methanol in the water to prevent microorganism growth.
16. Use a waste container to collect waste. Ensure the safety precautions to prevent spillage and evaporation in the lab.
17. Before starting the standard, verify the baseline.
18. Before starting the sample, verify the peak shape of standard, system suitability and area.
23. What is the meaning of Peak in chromatography?
Peak is a detector response represented as a chromatogram. One peak represents one component when separation is done adequately. When there is incomplete separation, i.e. two or more components’ peak gets merged because they eluted in overlapping manner and remain unresolved.
24. What is a Chromatogram?
Chromatogram a representation of detector response or a quantitative measurement value in a graphical form vs. the volume or time. Typical chromatograms have series of Gaussian peaks on a baseline.
25. What is Retention time (tR) in liquid chromatography?
Retention time, (tR) is the time between the injection of the compound and the maximum peak response of the eluted.
26. What is Dwell volume (D) in chromatography?
The volume between the is the volume between the point at which the eluents (mobile phase/ solvent) meet and the inlet of the column (Or the top of the column).
Dwell volume is also known as “gradient delay volume”.
27.What is Hold-up time (tM) in chromatography?
The time required for elution of an unretained component (In following schematic, an air or unretained solvent peak, with the baseline scale in minutes).

Reference: Working document QAS/21.905 – February 2022 (draft), 1.14.1 CHROMATOGRAPHY
28. What is Hold-up volume (VM) in chromatography?
The volume of mobile phase or solvent required for elution of an unretained component. It is
calculated using hold-up time ((tM)) and the flow rate (F), mL/min.
Formula is VM = tM × F
29. What is Number of theoretical plates (N) OR Plate number (N) in chromatography?
Number of theoretical plates (N) OR Plate number (N) is a measure that indicates column efficiency. In other words, it is a number that is indicative of performance of column OR column efficiency.
For Gaussian peaks, it is calculated by:
N = 16(tR/W)^2 (As per USP)
tR – retention time
W – peak width
For electronic integrators it can be determined using the equation:
N = 5.54 (tR/Wh)^2
(As per USP and Working document QAS/21.905 – February 2022 (draft), 1.14.1 CHROMATOGRAPHY)
tR – retention time
Wh – peak width at half-height (h/2).
30. What is Plate Height (H) in chromatography?
Theoretical plate height is height equivalent to a theoretical plate. It is a ratio of the column length (L), in micrometers, to the plate number (N): H = 𝐿/𝑁
It indicates the rate for the band broadening of peak broadening in HPLC equipment. The smaller the H value, the bigger the plate number.
When bigger the plate number, the better the column is packed and the smaller the dead volume of the instrument and, the sharper the peaks. It also means that the efficiency of the column is good.
31. What are the factors affecting the Number of theoretical plates (N) or Plate Height (H)?
- Substance being chromatographed
- Operating conditions i.e flow rate and temperature of the mobile phase or carrier gas
- Quality of the packing material
- The uniformity of the packing within the column
- For capillary columns, the thickness of the stationary phase film
- Internal diameter and length of the column
32. What is the meaning of Resolution (RS) in chromatography?
The resolution is the separation of two components in a mixture.
It is calculated using following formula:
RS = 2 × (tR2 − tR1)/(W1 + W2)
tR1 and tR2 are the retention times of the two components
W1 and W2 are the corresponding widths at the bases of the peaks obtained by extrapolating the relatively straight sides of the peaks to the baseline.
For electronic integrators it is determine using following formula:
RS = 1.18 × (tR2 − tR1)/(W1,h/2 + W2,h/2)
33. What is the meaning of Retention volume (VR) in chromatography?
Retention volume (VR) a volume of mobile phase or solvent required for elution of a component.
It is calculated using formula VR = tR (retention time) × F (flow rate in mL/min)
34. What is Peak-to-valley ratio (p/v) in liquid chromatography?
The peak-to-valley ratio can be considered as a system suitability criterion for related substances test in the condition of non achieving separation of two peak at the baseline level. Schematic representation is given as follows.


35. What is Relative retention (r) in liquid chromatography?
Relative retention (r) is the ratio between the adjusted retention time of a component relative to that of another used as a reference obtained under same chromatographic conditions.
It can be calculated using formula r = (tR2 − tM)/(tR1 − tM)
tR2 – retention time measured from the point of injection of the compound of interest
tR1 is the retention time measured from the point of injection of the compound used as reference
tM is the retention time of a non retained marker defined in the procedure
36. What is Relative retention time (RRT) in liquid chromatography?
Relative retention time (RRT) is also known as the “unadjusted relative retention”.
It is calculate using formula RRT = tR2/tR1
37. What is “Symmetry Factor” or “Tailing Factor” in liquid chromatography?
The Gaussian form of peak is generally completely symmetrical. When the rear is spread out to more or less extent and forms a ‘tail’. This phenomenon is called ‘tailing’.
“Symmetry Factor” or “Tailing Factor” (AS) is calculated using formula: W0.05/2f
W0.05- width of the peak at 5% height
f is the distance from the peak maximum to the leading edge of the peak
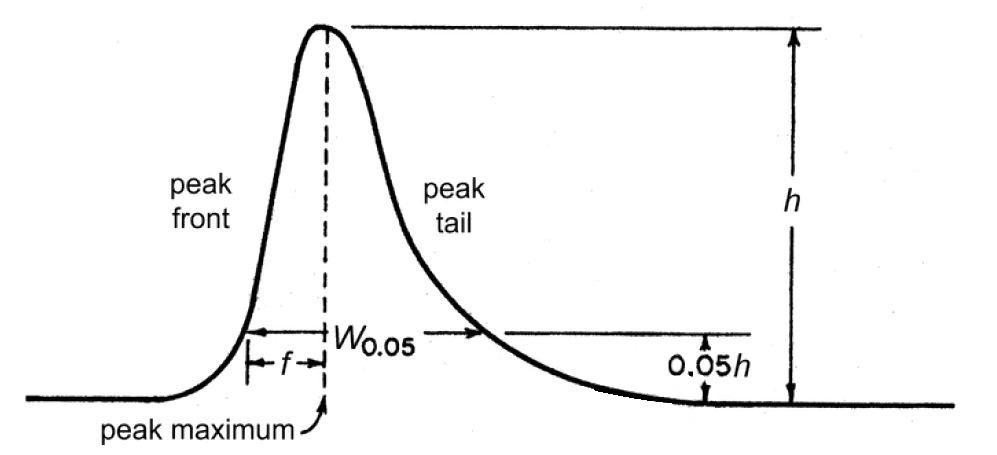
Reference: USP
38. What is ‘fronting’ or ‘leading’ in liquid chromatography?
In opposite of tailing phenomenon; when the front is flatter than the back, it is called as
‘fronting’ or ‘leading’.

39. What is System repeatability in liquid chromatography? OR What is System Suitability?
The repeatability and reproducibility of signal/ area/ response that is represented as % Relative Standard Deviation (% RSD) of consecutive measurements for Not Less Than (NLT) three standard solutions or applications of an applicable reference solution.
Total mobile phase time (tt) 279
In size-exclusion chromatography, the retention time of a component whose molecules 280 are smaller than the smallest gel pores (Figure 5).
40. What is the total mobile phase volume (Vt)?
This phenomenon is applicable for size-exclusion chromatography. The retention volume of a compound for those molecules having smaller than the minimum gel pores.
It may be measured using the flow rate (F) in ml/min and total mobile phase time. The equation is 𝑉𝑡=𝑡𝑡×𝐹
41. What is an ideal criteria for peak symmetry?
If not specified in the monograph or test method such as assay, related substance or other test, the symmetry factor or tailing factor applicable for the peak during quantitative evaluation is 0.8 to 1.8.
42. What is a signal-to-noise ratio?
The signal to noise ratio represents the capability of a method to detect or quantify the elute in a consistent manner; in other word, the signal-to-noise ratio represents the system sensitivity.
When the signal-to-noise ratio is more than 3, compounds can be consistently detected for each time. When the signal-to-noise ratio is more than 10, compounds can be consistently quantified for each time
43. What are the issues if the Reverse Phase HPLC column (stationary phase) is not cleaned properly?
Inadequate cleaning of the Reverse Phase HPLC column (stationary phase) results in a high back pressure, decrease in separation performance, broadening of peak, peak tailing and sometimes “ghost peaks” are detected.
This happens because of hydrophobic organic molecules, for example, lipids or large organic molecules easily stuck to RP HPLC Columns.
44. What is the procedure to clean the Reverse Phase HPLC column (stationary phase) quickly and efficiently?
Inject 100 to 200 microliter methanol or acetonitrile. Repeat this procedure 2 to 3 times. If a garbage peak still observes, do the normal flush with methanol or acetonitrile.
If you are using acetonitrile, ensure that it does not cause a precipitation of buffer-containing eluents.
45. What happens when the mobile phase is not degassed?
Noisy or drifting baselines and pressure fluctuations are signs of insufficient degassing.
46. What are the various procedures used for mobile phase degassing?
• Refluxing
• Vacuum degassing
• Helium degassing
• Ultrasonic degassing
47. What are the suggested procedures for mobile phase degassing?
• Refluxing – Method is good but it is not practicable.
• Vacuum degassing – Method is good.
• Helium degassing – Method is good.
• Ultrasonic degassing – Ineffective method. Works good for acetonitrile/ water mixtures.
48. What are the advantages of acetonitrile in the mobile phase?
- Low viscosity and better kinetics provides sharper peaks.
- acetonitrile/water mixtures have lower back pressure in comparison to methanol/water mixtures, hence, less wear and tear on seals and columns.
- Higher elution strength, henace, lower solvent consumption. It provided similar elution strength at a lower concentration compared to methanol.
- Silica gel is less prone to dissolve in acetonitrile compared to methanol (Reason: acetonitrile is less polar than methanol)
- Low solubility of air, hence, less problems with air and easy for effective degassing.
- Low UV absorptivity hence, better for detection at 195-200 nm
- Better reproducibility, specifically while using ionic solutes because it causes small pH deviations in aqueous solutions.
- Suitable for ion chromatography because it is a better solvating agent.
- More toxic than methanol resulting in prevention of microbiological growth in the instrument.
- Suitable for the separation of bases at lower pH and yield sharp peaks
49. What are the advantages of methanol in the mobile phase?
- Methanol is odorless hence, it provides better working conditions.
- Less toxic compared to acetonitrile.
- Better solubility for salts hence, chances of precipitation low.
- Methanol/water mixture brings the seals faster into its swelling conditions hence, equipment gets faster to working condition
- On aging of acetonitrile, impurities such as propionitrile, methacrylonitrile generate ghost'” peaks. This issue is less known for methanol hence, longer shelf life of methanol.
- Good separation of bases at alkaline pH
- Lower baseline noise above 220 to 230 nm
50. What could be the impact of too high or too low pH of the mobile phase on the C18 column?
Impact of high pH environment on C18 Column is silica gel dissolves at above pH 8. Because of this, the column performance will rapidly decrease.
Impact of low pH environment on C18 Column, C18 chains are hydrolyzed at pH 2 and lower. Because of that, the column bleeds, and the performance decreases.
51. What is the solution to deal with very high (above pH 8) or very low pH (at pH 2 and lower) mobile phase?
Install a pre-column or old CI8 column between the pump and the injector.
On installation of this precalumn, the alkaline eluent will get saturated with silica gel and will not affect the separation column.
At low pH, the C18 chains of the pre column are hydrolyzed, protecting the main column.
52. What are the possible reasons for a change in retention time in HPLC analysis?
Following are potential reasons for change in retention time in HPLC analysis.
Change in stationary phase
Change in mobile phase
Change in temperature
Change in flow rate
Change in packing density
53. What are the most probable reasons for the short life span of the HPLC Columns?
- Strongly adsorbed contaminants in the sample can ruin the column performance
- Shedding seals may clog column filters and the top layers of the packing.
- Mechanical weakness of the packed bed which could be the consequence of rough handling of the column while handling in the lab or during shipment..
- Inconsistent column life may occur because of adsorption of sample constituents on the top of the column. This could either occur because of precipitation because of low solubility of sample in the mobile phase or they may be strongly adsorbed. This can also happen if one injects more samples. In that case, contaminants build up on the top of the column and prevent the sample to properly adsorb and distribute. This issue results in a peak distortion and an increase in back pressure.
- Column collapsing may occur if a mobile phase pH outside the recommended range of column.
- Sample dissolved in a strongly acidic or alkaline solution.
- Columns exposed to the wrong solvents.
- Repeated column “washing”.
54. What are the solutions to increase the life span of the HPLC Columns?
- Sample clean up with a suitable sample preparation method.
- Use a guard column or precolumn. This will prevent column clogging and accumulation of contaminants at the top of the column. To have the highest performance of the guard column, use exactly the same packing as the analytical column.
- (Not preferred method) Column washing could be one of the solutions that dissolves the contaminants on the top of the column. The drawback is washing will remove hydrolyzed bonded phase. Therefore, a repetitive washing may result in accelerated aging of the column.
- (Not preferred method) Column backflushing. The drawback is when washing with a different solvent than the mobile phase, same consequences as column washing. While doing this using the mobile phase, it’s time consuming and sometimes it doesn’t work. (Note: Backflushing should not be made as standard practice).
55. What are the possible reasons for variable Retention Times (RT) in HPLC analysis?
Randomly changing and inconsistent retention times – Pump(s) and the solvent mixing devices malfunction and not giving consistent flow.
- Retention time does not change from run to run but they vary from day to day – most likely the source of variation is the composition of the mobile phase. Note: Rule of thumb, if you make an error of 1% in the amount of organic solvent, the retention time can change by between 5% and 15%, typically by about 10%.
- Temperature fluctuations (Rule of thumb – RT change by about 1% – 2% per 1 ºC).
- Air conditioning shut down usually at weekends.
56. What are the solutions to prevent the variation in retention time in HPLC analysis?
- Accurately preparation of the mobile phase. While preparing the mobile phase, measure the amount of solvent very carefully. Preferred method to prepare the mobile phase gravimetrically rather than volumetrically.
- Method of degassing of mobile phase contributes to variability. Very good degassing method is applying vacuum and ultrasound together for about one minute. Consequently there will be very good degassing with a nominal amount of solvent evaporation. Another good method is the use of helium sparging. Precaution: After initial equilibration, the flow of helium to be reduced to prevent helium to carry solvent vapors with it and reduce evaporation.
- Accurate measurement of pH. A change of as little as 0.1 pH units can result in a retention time shift of 10%.
- Control of the pH is critical when your sample contains ionic or ionizable compounds.
- Temperature is an important factor to prevent the fluctuation of retention time.
57. What are the reasons for Drifting Retention Times in HPLC analysis?
Most common cause of drifting retention times is an equilibration problem.
In normal phase chromatography the Retention time is sensitive to the quantity of water adsorbed on the silica surface. Solubility of water in solvents such as methylene chloride or hexane is very low, column equilibration takes time. Still there are chances that RT my shift when dry hexane is used.
Equilibration in reversed-phase chromatography is quicker; 5 to 10 column volumes of mobile phase is generally sufficient to equilibrate column.
58. What is the solution for prevention of drifting Retention Times in HPLC analysis?
As we understood, the most common cause of drifting retention times is an equilibration problem.
In normal phase chromatography it is recommended to avoid using very dry solvents. Most preferable approach to solve the equilibration problem of silica with water is to use solvents that are “half-saturated” with water. It can be prepared by saturating a provided volume of hydrophobic solvent with water and mixing it 1:1 with “dry” solvent. This will facilitate quick equilibration of the column.
In reversed-phase chromatography, 5 to 10 column volumes of mobile phase are generally good for quick equilibration.
59. What are the possible reasons for the peak shape issue of Peak Tailing?
- Wrong mobile phase pH
- Column void
- Blocked frit
- Unswept dead volume
- Interaction with active silanols
- Chelation with metal ions in stationary phase
60. What are the solutions to prevent peak shape issues of Peak Tailing?
- Wrong mobile phase pH → Increase buffer concentration or Decrease mobile phase pH that suppress silanol ionization
- Column void → Change the column, Column backwash
- Blocked frit → Column backwash, Use inline filters
- Unswept dead volume → Use shorter tubing connection and minimum number of connection, Ensure tightening of all connections
- Interaction with active silanols or Chelation with metal ions in stationary phase → Add basic mobile phase additive or Use ultra-high purity silica based stationary phase
61. What are the possible reasons for the peak shape issue of Split Peaks?
- Contamination on column inlet
- Incompatibility of Sample solvent with mobile phase
- Elution of second component simultaneous
- Blocked frit
- Column overloaded
62. What are the solutions to prevent peak shape issues of Split Peaks?
- Contamination on column inlet → 1. Use guard column, or replace guard column, or replace analytical column (depending on issue identified), 2. Backwash analytical column (less preferred method and should be rarely used), 3. If contaminants are strongly adsorbed, try to regenerate column Or 4. Replace column if issue is not resolved
- Incompatibility of Sample solvent with mobile phase →
- Elution of second component simultaneous → 1. Sample cleanup before injection, 2. Modify mobile phase composition, or 3. Change stationary phase depending on selectivity
- Blocked frit → Use in-line filter, Column backwash
- Column overloaded →1. Increase column capacity by using high capability stationary phase or increase the dimension, or, 2. Decrease injection volume
63. What are the possible reasons for the peak shape issue of Peak Fronting?
- Sample solvent incompatible with mobile phase
- Formation of channels in column
- Low temperature of column oven
- Column overloaded
64. What are the solutions to prevent peak shape issues of Peak Fronting?
- Low temperature of column oven → Increase column oven temperature to increase column temperature
- Column overloaded → Use less injection volume or dilute the sample solution according to desired column load
- Formation of channels in column → Refer to the column literature to operate the system within the given range of parameters, e.g. pH of the solution. If problem persist, replace the column
- Sample diluent incompatible with mobile phase → Change the sample diluent as mobile phase
65. What are the types of Retention Time Variation or RT Variation?
- Decreasing Retention Times
- Increasing Retention Times
- Fluctuating Retention Times
66. What are the probable reasons for Decreasing Retention Times?
- High flow rate
- Column overloaded
- Loss of bonded stationary phase
- Active groups on stationary phase
- Incorrect column selection
67. What are the solutions to prevent the issue of Decreasing Retention Times?
- High flow rate – Check and regulate the flow rate of pump
- Column overloaded – Decrease sample injection volume, Select the column with larger internal diameter
- Loss of bonded stationary phase – Replace column, Operate at recommended pH range for RP columns. Generally operated at 2-8 for silica based RP HPLC.
- Active groups on stationary phase – Increase buffer strength
- Incorrect column selection – Select the column with larger internal diameter
68. What are the probable reasons for Increasing Retention Times?
- Changing mobile phase composition
- Low flow rate
- Loss of bonding in stationary phase
- Bubbles in mobile phase
69. What are the solutions to prevent the issue of Increasing Retention Times?
- Changing mobile phase composition – 1. Cover solvent reservoirs, 2. Prepare fresh mobile phase
- Low flow rate – 1. Check and adjust pump flow rate, 2. Check for leaks in system, including pump seals
- Loss of bonding in stationary phase – Replace column
- Bubbles in mobile phase – 1. Check flow rate and pressure, 2. Degas mobile phase
70. What are the probable reasons for Fluctuating Retention Times?
- Inadequate column equilibration
- Mobile phase preparation error/ composition variation
- Inadequate buffer capacity
- Unstable column temperature
71. What are the solutions to prevent the issue of Fluctuating Retention Times?
- Unstable column temperature – 1. Check column oven to ensure temperature stability, 2. Malfunctioning of the column oven thermostat
- Inadequate column equilibration – 1. Allow columns to equilibrate for sufficient time between runs, 2. Condition the column using concentrated sample
- Mobile phase preparation error/ composition variation – 1. Verify volume make-up of mobile phase, if needed, prepare fresh one, 2. Verify the proportioning-valve accuracy
- Inadequate buffer capacity – Utilize the buffer concentrations more than 20mM
72. What are the probable reasons for Ghost Peaks?
- Contamination of column
- Contamination of injector
- Carryover/ late eluting peak from previous injection
- Contaminated water or solvent
- Method specificity not established adequately
73. What are the solutions to prevent the issue of Ghost Peaks?
- Contamination of column – Adequate column washing/ Flushing of column to remove contaminants
- Contamination of injector – Injector flushing between analyses
- Carryover/ late eluting peak from previous injection – 1. Increase the run time, 2. Flush column with strong mobile phase at end of each run, 3. For gradient runs, end the run at higher concentration
- Contaminated water or solvent – Use HPLC grade water or solvent
- Sample contamination – Ensure cleanliness of glasswares, sample preparation aids such as mortar and pestle, cleanliness of sample storage area
- Method specificity not established adequately – Use sample clean-up process and perform method specificity covering all the relevant factors (excipient peak interference, degradants, etc.)
74. What are the probable reasons for Negative Peaks?
- When using RI detector, Refractive Index of solute is less than the mobile phase
- When using UV detector, solute absorption is less than absorption of mobile phase
- Different composition of Sample solvent and mobile phase
75. What are the solutions to prevent the issue of Negative Peaks?
- When using the RI detector, the Refractive Index of solute is less than the mobile phase – 1. Use mobile phase with less refractive index, 2. Invert detector polarity to get positive peaks
- When using a UV detector, solute absorption is less than absorption of mobile phase – 1. Change UV wavelength to get a positive peak, 2. Identify mobile phase so that it lowers the UV absorption
- Different composition of Sample solvent and mobile phase – Revise the composition of either sample solvent and or mobile phase to improve the compatibility and sample solubility
76. What are the probable reasons for Spikes in a chromatogram?
- Air bubbles in mobile phase/ inadequately degassing of mobile phase
- Column stored without closing ends using column closure caps
77. What are the solutions to prevent the issue of Spikes in a chromatogram?
- Air bubbles in mobile phase/ inadequately degassing of mobile phase – 1. Degas mobile phase using suitable method, 2. Use back pressure restrictor at detector outlet, 3. Ensure all tubings are without any leaks and fittings are tight
- Column stored without closing ends using column closure caps – 1. Ensure that column is stored with end caps closed, 2. Flush Reverse Phase column using degassed methanol
78. What are the probable reasons for High Back Pressure in HPLC systems?
- Wrong HPLC pump setting
- Pressure higher during middle of gradient
- Temperature lower than required
- Column ageing
- Column frit blockage
- In-line filter blockage
- Guard column blockage
- System blockage
- Buffer precipitation in the system
79. What are the solutions to prevent the issue of High Back Pressure in HPLC systems?
- Wrong HPLC pump setting – Verify HPLC pump setting and adjust appropriately
- Pressure higher during middle of gradient – This is normal phenomenon
- Temperature lower than required – Maintain the column oven temperature as per method requirement
- Column ageing – It is normal phenomenon of gradual increase in pressure over the period of lifetime
- Column frit blockage – 1. Column backwash, 2. Use an in-line filter to prevent column blockage and reduce backpressure, 3. Ensure efficient filtration, 4. Use guard columns
- In-line filter blockage – 1. Replace in-line filter frit with new one, 2. Centrifuge or filter samples with recommended filters, 3. Degas and Pre-filter mobile phase
- Guard column blockage – Increase the replacement frequency depending on experience
- Buffer precipitation in the system – Back flush the column using water
80. What are the probable reasons for Low Back Pressure in HPLC systems?
- Leakage in the HPLC system
- Column temperature is higher than required
- Flow rate is lower than required
81. What are the solutions to prevent the issue of Low Back Pressure in HPLC systems?
- Leakage in the HPLC system – Identify the leakage and correct it
- Column temperature is higher than required – Set the temperature as per method requirement
- Flow rate is lower than required – Set the flow rate as per the method requirement
82. What are the good habits that help to minimize the HPLC system related issues such as peak shape problems, retention time variation, ghost peaks and column back pressure issues?
- For new projects, select well researched, high-purity silica-based column and use highest quality HPLC-grade reagents.
- Flush the HPLC system at regular intervals that removes salts and buffers.
- Service the system periodically to reduce check-valve and pump-seal problems.
- Precise sample preparation with adequate filtration and sample clean-up process reduces sample related issues.
- Strong-solvent flush after every run or at specific frequency will reduce sample carryover and extend column lifetimes.
- Columns won’t last forever, but with proper care, you should be able to get a good return on your investment.
Refer Complete guide on High Performance Liquid Chromatography (HPLC)