

White paper on Prevention of Contamination and Cross-Contamination in shared Pharmaceutical Manufacturing Facilities
Introduction
Manufacturing pharmaceutical products involves various unit operations. The equipment, utility, facility, and other supporting systems are used in a multi-product manufacturing facility to carry out unit operations.
Contamination or Cross-contamination could be an outcome of accidental mix-up of starting material, intermediate, or finished product with foreign material, with other products’ starting material or other product. The contributory factors for contamination or cross-contamination could be human, facility, procedure, equipment, or utility. The contamination and cross-contamination could occur at any stage, starting from raw material sources to the packaging of pharmaceutical products.
The contamination or cross-contamination could occur during:
• Material transportation and receipt at a manufacturing facility
• Storage
• Sampling
• Dispensing
• Manufacturing
• Packaging
The white paper provides general guidance to detect the source of contamination and cross-contamination and mitigation strategy with the help of facility design and procedural controls.
The white paper provides general guidance to detect the source of contamination and cross-contamination and mitigation strategy with the help of facility design and procedural controls.
References:
• EU GMP Chapter-3 (Premises and equipment)
• EU GMP Chapter-5 (Production)
• 21 CFR Part 211: Subpart C: Buildings and Facilities 211.176
• ISPE’s risk map Guide to managing Risks Associated with Cross-Contamination inline with Chapter-3 and Chapter-5 of EU GMP.
• ICH Q9: Quality risk management Penicillin Contamination
Key attributes to contamination and cross contamination
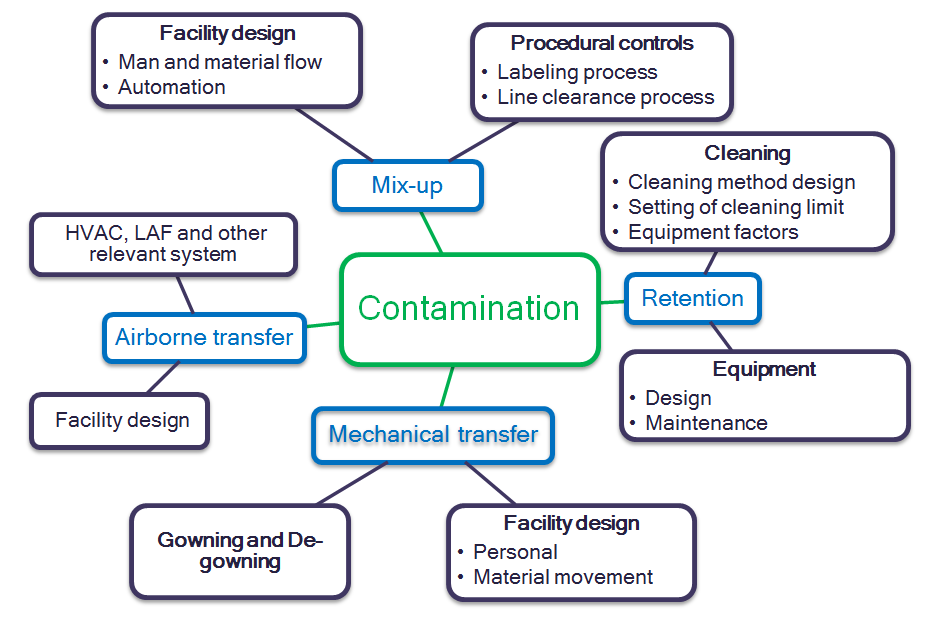
According to the ISPE Baseline Guide Vol. 7 – Risk Based Manufacture of Pharmaceutical Products, Section 6.3, the main contributory factors for contamination and cross-contamination are Mix Up, Retention, Mechanical Transfer, and Airborne Transfer. Therefore, these key attributes can be considered during the Quality Risk Management process of a shared pharmaceutical product manufacturing facility.
The outcome of the Quality Risk Management process should be the basis for determining the extent of technical and organizational measures required to control risks for cross-contamination. For example, cross-contamination can be prevented by the robust design of the facility, equipment, utility, material movement, personnel movement, manufacturing procedure, and cleaning procedures within a manufacturing facility.
Figure 1: Sources of Contamination and Cross- Contamination
Prevention of contamination and cross-contamination
A. Mix-up
Mix-up is a contamination of the product by combining various compounds or mixing one product or material with another. The mix-up could result from inadequate facility design, the inadequate flow of material, poorly defined procedures, or human error.
a. Facility design
● Designated storage areas should be provided for:
→ Quarantine materials
→ Approved materials
→ Rejected materials
→ Dispensed material
→ In-process materials ( Example: compressed tablets and coated tablets)
● The different areas should be provided for clean equipment storage and unclean equipment storage.
● Lock and key arrangement should be provided for printed packing materials.
● The area should be designed for adequate room differential pressure in the manufacturing facility: manufacturing rooms, clean equipment rooms, unclean equipment rooms, etc.
● A dedicated facility should be provided for high potent molecules, or single-use disposable technologies should be used.
● The facility should be designed for unidirectional personnel and material flow. Material includes raw materials, in-process materials, packing materials, finished goods, and scrap.
● Authorized personnel movement and access control to prevent unauthorized access.
● Adequate lighting should be provided to carry out operations.
● Automated control can be deployed for material verification and segregation, such as barcoding or similar system.
● The visual inspection system can be implemented for product inspection, packed goods inspection to verify the overprinting matter. These provisions can be part of the track and trace system.
● Dust collectors should be interlocked with Air Handling Unit (AHU). Non-return valve at user point of dust extraction system to prevent backflow when power outage.
b. Procedural controls
i. Labeling process
● Implementation of the labeling process starts from material receipt to goods distribution. For example:
→ Material receipt
→ Storage in warehouse – quarantine, under test, approved, rejected, etc.
→ Dispensed material
→ In-process quarantine
→ Online rejections
→ In-process rejects
→ Under maintenance
→ Clean equipment
→ Unclean equipment
→ Area status label
● Archival of cleaning labels with the batch record.
● Training on the labeling process.
● Procedure for authorization to access control systems.
● Procedure for signboard.
ii. Line clearance process
● A procedure for line clearance should be implemented to check the cleanliness of the area and equipment between the batches and products. Doer and checker principle should be in place for area clearance.
● The visual verification process should be available to ensure the cleanliness of equipment and area, emphasizing hard-to-clean and hard-to-reach areas.
● Equipment cleaning verification can be done through a swab or rinse sampling for the product with low Permitted Daily Exposure (PDE) value after product changeover and before starting of next product batch.
● Product dedicated equipment should be used for high potent molecules.
● Product dedicated equipment accessories can be used for hard-to-clean surfaces on which low PDE value products are getting manufactures—for example, sieves, screens, chequered plates for pallet manufacturing.
● Visualization aid should be used to enhance the visibility of previous product residue (Example of visualization aid are a torch, mirrors), or indirect methods can be used (such as black and white cloth) to ensure the absence of previous product traces after cleaning equipment.
iii. Other procedures which can be adequately designed to prevent mix-up are:
● Personnel and material movement.
● Scrap management.
● Physical separation of the high risk products.
● Access controls.
● Visual inspections.
● Material segregation and handling.
● Facility, equipment, and utility qualification, periodic re-qualification and routing monitoring.
● Personnel qualification and training.
● Procedure for identification of high potent molecules.
● Procedure for sign board, area labeling, personnel and material flow directions, and utility identification.
B. Retention
When the manufacturing facility uses shared product contact equipment surfaces to manufacture the different products, there is a possibility of carryover of material of one product to subsequent product manufactures on same equipment.
The risk of retention could be the contribution of inadequate cleaning due to the nature of the material, inadequate cleaning procedure, or non-adherence to the cleaning procedure.
a. Cleaning
i. Cleaning method design
● The preferable method for equipment cleaning is the automated cleaning process because it reduces the chances of variation due to human intervention — for example, Clean-In-Place (CIP) system. The CIP system should be recipe-based and finalized for use after validation.
● When using a manual cleaning process, cleaning methods should be developed to reduce person-to-person variability. This can be achieved by optimizing the variables contributing to the cleaning process. For example, when using water as a cleaning aid, water temperature range, water pressure, water spray nozzle diameter, time for spraying the water, the quantity of water to be used, specifying the parts to be cleaned, the concentration of cleaning agent (if used).
● For new products, R&D should provide the equipment cleaning process based on their experience during the product development stage.
● Reproducibility of the cleaning process can be proven by validating the cleaning process by manufacturing different batches, and products and using different operators for different batches.
● Dirty equipment hold time study and cleaning efficiency after campaign should be evalauted to ensure the effectiveness of cleaning process.
● Cleaning of equipment until visually clean is not an acceptable process of cleaning.
● Identify hard-to-clean parts of equipment during cleaning method development and specify them in the cleaning procedure.
● Filters, fluidized bed processors bags, and venting bags of granulator should be product dedicated because prevention of residue of previous product to the following product cannot be guaranteed.
● Cleanliness verification should be done by at least two different personnel for cleaning adequacy.
● Development of visual cleanliness criteria to declare cleaning of equipment as visually clean.
● Visualization aid should be used to verify equipment cleanliness. Examples are a torch, mirror, etc.
● The method used to measure the residue of the previous product after cleaning should be capable to accurately and precisely determine the acceptable residual level. The method used to determine the residue after cleaning must be validated as per guidance on analytical method validation.
● The operator should be trained on the relevant cleaning procedures.
ii. Setting of cleaning limit
● The setting of the cleaning limit shall be based on the scientific sound rationale to prevent the cross-contamination of the subsequent product by the previous product.
● Preliminary assessment of cleaning should be done using visual cleaning criteria.
● Health-Based Exposure Limit (HBEL) should be considered to determine the cleaning limit.
● Toxicology data should be used to determine the Permitted Daily Exposure (PDE) limit, which can be further used to calculate the cleaning limits.
● Factors to be considered while determining the criteria for residues should be toxicity, potency, and sensitivity or allergic reaction.
● Criteria for worst-case product selection should be Solubility, Cleanability, and Toxicity.
iii. General housekeeping
● Procedure for the handling of material spillage should be established.
● In a high dust generation area, the procedure for intermittent cleaning should be implemented to reduce the amount of dust in the area.
● Area and floor cleaning should be up to the adequate level that it should not generate dry residue of the previous product once the area is dry after cleaning.
● Only trained person should perform housekeeping activity in the area.
b. Equipment
i. Design
● Material of construction (MOC) of equipment product contact part should be non-adsorptive, non-reactive, non-porous, corrosion-resistant, smooth, non-releasing, and surface should be easily cleanable.
● Majorly used product contact surfaces are stainless steel SS316 and SS316L, food-grade plastic and rubber, non-leachable glass, etc.
● The product contact surface of the equipment should be mirror-finished so that it should be easily clean.
● Equipment should be designed in such a way that all the parts of the equipment are easily accessible and visible to ensure adequate cleaning and cleanliness of the equipment.
● Whenever piping is involved in the equipment design, which is the product contact part, ensure that joints are adequately welded. Inert gas and orbital welding should be used, followed by boroscopy of the piping to ensure smoothness of the surface. The dead leg should be less than 2D. Drainability of piping should be ensured.
● Ensure the dent-free surface of the product contact part of the equipment.
● Dedicated sampling tools can be used for low PDE value product sampling.
ii. Equipment maintenance
● Equipment should be periodically inspected to check the presence of scratches, dents, and cracks that may impact the cleanability of equipment.
● Procedure for periodic equipment maintenance should be in place to ensure consumable replacement such as gaskets, valves, view glass, lids, tri-clover joints, etc., based on its condition.
● The above activity should be adequately documented and part of the approved procedure.
● Equipment should be evaluated for its retirement based on the trend of breakdown and maintenance history.
● Depending on the nature of the process, equipment should be product dedicated to minimizing the risk.
C. Mechanical transfer
The mechanical transfer of contaminants is the transfer of contaminant from non-contact product surfaces, such as equipment, accessories, area, gowning, etc., into the product.
The mechanical transfer could be the source of contamination when the personnel and material flow is inadequate because of the poorly designed facility or procedure. Gowning and protective clothing also play a role in cross-contamination when handled incorrectly or poorly designed procedures.
a. Facility design
i. Personal movement
● There should not be a cross-flow of exposed material or personnel between rooms where different products are being manufactured or between dedicated product manufacturing facilities.
● For manufacturing highly potent drugs or sensitizing molecules, the entire facility, including walkways, washrooms, and canteen, should be separate.
● While personnel are moving to the common passage, de-gowning should be done.
● While dispensing of material, personnel and material movement should be done in the designated area.
● The washroom should be outside the manufacturing area’s main entry. Gowning and de-gowning procedures should be followed while accessing the washrooms.
● Provision of interlocking of doors of change rooms should be provided. Strict adherence to the change room process by the staff is essential.
ii. Material movement
● While the movement of material from one place to another place, it should be in closed condition.
● Material container and transfer trolley should be adequately cleaned so that it will not carry the residue of product to other areas.
● Material movement trolley wheels should be cleaned before they come out from the process rooms to prevent residue carryover into the common corridor or other areas.
● The material should be moved from one area to another area as per the material movement path only.
● Scrap should be moved adequately in the closed condition. For example, while taking out the scrap bag from the processing room, it should be enclosed in the second bag to prevent residue carryover to the scrap movement path.
b. Gowning and de-gowning
Gowning is the process of using special clothing in clean rooms to control contamination. The types of gowning required are depending on the nature of work, area classification requirement. There are two purposes of gowning; to protect people from exposure to unwanted chemicals and protect the product.
Gowning covers head/ hair, beard/ mustache, face, torso/ upper body, legs/ pants, feet/ shoes, and hands. The gowning is designed and intended to cover all necessary parts of the body to prevent hair, skin cells, and other particles on oneself from shedding into the cleanroom environment and/ or contaminating the product. Therefore, the importance of gowning should be implied.
● Entry to clean rooms should be done only through the personnel entry with proper gowning.
● Gowning should be done within the designated area in the change room.
● The sequence of gowning should be properly followed for correct gowning.
● Depending on operations, Personnel Protective Equipment (PPE) should be part of the gowning procedure.
● Wearing jewelry, wristwatch, nail polish/ false fingernail, cosmetics, hair spray, perfumes should be restricted, which should be part of the approved procedure.
● The gowning should be maintained in the cleanroom throughout the duration up to which person stays inside the area.
● Personnel hygiene is one of the critical components. It includes but is not limited to Shower, Shave, Brush teeth, Brush hair, etc.
● Street clothing and shoes must not be worn within GMP areas.
● Steps for de-gowning should be part of the procedure.
● After de-gowning while leaving the cleanroom, it should be kept in a designated place to prevent contamination in the change room.
● In case of entry to the aseptic area, the competency of gowning/ de-gowning should be assessed through monitoring by experienced and qualified staff, followed by microbiological testing.
● The quality of cleanroom clothing should be designed so that it should not shred particles. The gowning should withstand repeated wear and laundering.
● Direct contact between the operator and starting materials, primary packing materials, and intermediate/ semi-finished or ready-to-pack products should be avoided.
● Fresh gowns should be used daily.
● For the aseptic area, sterile gowns should be used.
● For low PDE value or high potent drug manufacturing, disposable/ dedicated gowns should be used.
● While collecting the gowns at the end of the day to send for the washing, they should be segregated to prevent cross-contamination.
● Procedure for gown collection, segregation, washing, maintenance, and management of clean gown should be well established to ensure that the same process is being followed every time.
D. Airborne transfer
Air is a carrier of powder dust or material aerosol and transfers it from one place to another place. Therefore, uncontrolled or unprocessed air movement in the facility may pose risk of contamination or cross-contamination in the pharmaceutical product manufacturing facility.
Atmospheric unprocessed air has pollutant have high potential to contaminate the product. Similarly, when air with powder dust transfers from one processing area to another processing area, it can cause cross-contamination. Poorly designed facilities and utilities can cause airborne contamination or cross-contamination of pharmaceutical products.
a. HVAC, LAF and other relevant system
i. HVAC System
● The HVAC system should be used to filter the supplied air to prevent airborne contamination from the atmosphere.
● Air supplied to the classified area should have adequately designed Heating Ventilation, and Air Conditioning (HVAC) system where air should be supplied through series of filters such as primary filter (For example, 10 µ filter), secondary filter (For example, 5 or 3 µ filter), and final air filtration should be through High-Efficiency Particulate Air (HEPA) filters with 0.3µ.
● The preferable approach is to have an area dedicated Air Handling Unit (AHU) where the same AHU should not be supplied to areas where different products are being manufactured to prevent cross-contamination.
● The filters should be monitored using Differential Pressure (DP) indication to ensure that filters are not torn or clogged. HEPA filters should be periodically monitored for the leak.
● Adequate recirculation should be done so that the ratio of fresh air to recirculated air is maintained.
● Adequate differential pressure should be maintained between the area and between classification zones to prevent cross-contamination.
● For the Oral Solid Formulation facility, the corridor should be maintained with positive pressure than the processing area to prevent cross-contamination.
● For a sterile formulation facility, processing areas should be maintained with positive pressure with respect to the corridor to prevent cross-contamination.
● Primary and secondary filters should be cleaned at a defined frequency.
● The design of the dust extractor should enable to extract dust from the area. Dust extraction system air should not return dust into the production area. Dust extractor should be interlocked with the AHU unit to prevent backflow.
● Wash area air should not be taken into AHU return to mitigate the potential for contamination.
● Air-handling systems for the manufacture, processing, and packing of penicillin shall be completely separate from those for other drug products for human use.
ii. Reverse Laminar Air Flow (RLAF) station
● Dispensing of material should be done under RLAF to contain the dust of raw material to prevent the potential for cross-contamination.
● The preferable approach is to have separate RLAF for API and Excipient. However, the same RLAF can be used where dispensing of active material should be done at the end.
● RLAF should be validated to ensure that airflow is upward to downward towards the return air filter and room air is not getting mixed with air under RLAF airflow. The airflow can be visualized using a smoke study (Air Flow Visualization study) to ensure that the RLAF is working as intended.
● A safe zone should be defined in the RLAF with the help of a smoke study.
● The final filtration of air should be through a HEPA filter.
● After every changeover, RLAF should be stabilized to achieve the desired classification. The stabilization time should be established through validation studies. Stabilization time monitoring should be part of the line clearance procedure.
● Differential pressure across the primary, secondary, and HEPA filters should be monitored to ensure the effective functioning of RLAF.
● For the high potent or low PDE value product, dedicated filters should be used.
iii. Laminar Air Flow
● For sterile formulation facilities, Unidirectional (or Laminar) airflow systems should be used to manage contamination, particularly in Grade A areas with a low airborne particle limit. This is achieved by passing air through HEPA filters and directing it downward in a constant parallel stream towards filters located on walls near the cleanroom floor or through raised perforated floor panels, which are then recirculated.
● The filter cleaning procedure should be adequately defined.
● Primary and secondary filters should be cleaned as per frequency.
● Integrity/ restriction for these filters should be ensured based on the pressure gauge provided across the filters.
● HEPA filters should be verified for their integrity once a year for Grade C, D, and six-monthly for Grade A and B areas of the sterile formulation facility.
● For low PDE value and high potent drug products, primary filters should be product dedicated.
iv. Compressed air system
● Compressed air used during manufacturing of drug product and for cleaning should be finally filtered through 0.22 micron filter (0.01 micron air filtration).
● In a sterile formulation facility, a 0.22 micron filter should be used at the point of use.
● Periodic qualification of compressed air should be carried out to verify that it is meeting the standards.
● Supplied air should be dry enough so that it will not produce condensate within the line.
● The material of construction of compressed air line should be non-corrosive to prevent any potential contamination.
b. Facility design
● Any building or buildings used in manufacturing, processing, packing, or holding a drug product shall be of suitable size, construction, and location to facilitate cleaning, maintenance, and proper operations.
● Adequate space should be available for the orderly placement of equipment and materials to prevent mix-ups between different components, drug product containers, closures, labeling, in-process materials, or drug products, and to prevent contamination.
● The flow of components, drug product containers, closures, labeling, in-process materials, and drug products through the building or buildings shall be designed to prevent contamination.
● Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable.
● Operations relating to the manufacture, processing, and packing of penicillin/ β-lactam antibiotic products should be performed in facilities separate from those used for other drug products for human use.
● Adequate lighting should be provided in all areas to perform operations adequately.
● Equipment for adequate control over air pressure, micro-organisms, dust, humidity, and temperature should be provided when appropriate for manufacturing, processing, packing, or holding a drug product.
● Potable water should be supplied under continuous positive pressure in a plumbing system free of defects that could contribute contamination to any drug product.
● Drains should be of adequate size and, where connected directly to a sewer, shall be provided with an air break or other mechanical device to prevent back-siphonage.
● Sewage, trash, and other refuse in and from the building and immediate premises should be disposed of in a safe and sanitary manner.
● Adequate washing facilities should be provided, including soap or detergent, air driers or single-service towels, and clean toilet facilities easily accessible to working areas.
● The building used in the manufacture, processing, packing, or holding of a drug product shall be maintained in a good state of repair.
● The building used for the manufacture, processing, packing, or holding of a drug product should be maintained in a clean and sanitary condition.
● The building should be free of infestation by rodents, birds, insects, and other vermin (other than laboratory animals).
● Written procedures should be used for suitable rodenticides, insecticides, fungicides, fumigating agents, and cleaning and sanitizing agents to prevent the contamination of equipment, components, drug product containers, closures, packaging, labeling materials, or drug products.