
Handling of variations to Marketing Authorisation in EU region

High level summary for basic understanding
When the pharmaceutical manufacturer deals with the European regulatory region to market and distributes medicines, understanding variations to Marketing Authorisation concerning post-marketing change is essential. The knowledge of handling post-marketing changes is necessary for Quality Assurance specialists, Regulatory Affairs specialists, and Production Heads.
The post Marketing Authorisation changes are of various types and will undergo different types of approval processes. We will discuss the types of variations and classification; Type IA, Type IAIN, Type IB, Type II variations, and what Extension applications are. Furthermore, variations are broadly classified as Minor Variations (Type-1A & Type-1B variations) and Major Variations (Type-II variations).
1. What is a Variation….?
“A variation to the terms of a marketing authorization is an amendment to the contents of the documents of the approved dossier.” Variations in European Union (EU) are regulated by following regulations and guidelines:
Commission Regulation (EC) No. 712/2012 of 3 August 2012 amending Regulation (EC) No. 1234/ 2008 concerning examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products.
Annex I – Extensions of marketing authorisations
Annex II – Classification of variations
Annex III – Cases for grouping variations
Annex IV – Elements to be submitted
Annex V – Variations concerning a change to or addition of therapeutic indication, addition of non-food producing target species, replacement or addition of a stereotype, strain, antigen etc.
2. Categorization of changes in general
In general terms, the changes are categorized into three types depending on the nature of the category:
a. Administrative changes
b. CMC (Chemistry, Manufacturing and Controls) – Quality changes
c. Safety, Pharmacovigilance (PV), Efficacy etc.
3. Types of variations and approval requirements from the agencies
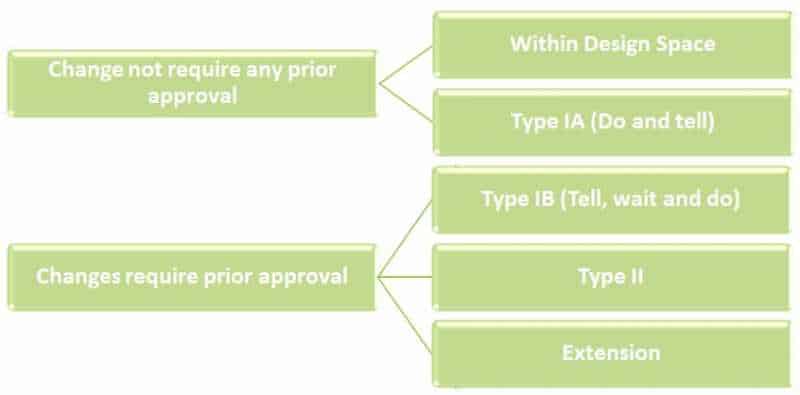
3.1 Types of variations are broadly classified in following types
Minor Variations: Type-1A & Type-1B variations
Major Variations: Type-II variations
Extension applications
3.2 Classification of variation with respect to types
Type IA – “Do and tell” for changes implemented in previous 12 months
Type IAIN – “Do and tell” and requires immediate notification after implementation
Type IB – “Tell, wait and do” and requires notification before implementation (wait 30 days after submission of procedure)
Type II – More detailed changes (“Tell and do”) and requires approval before implementation (usually 60 day review; range 30-90 days)
Extension applications – e.g. additional strengths, pharmaceutical form and route of administration (up to 210 day review)
4. Detailed explanation of all types variations to the Marketing Authorisation in EU region
4.1 Type IA variations
Type lA variations are the minor variations that have only a minimal impact or no impact on the quality, safety, or efficacy of the medicinal product, and do not require prior approval before implementation (“Do and Tell” procedure).
i. Type lA variations requiring immediate notification (‘IAIN’)
ii. Type lA variations NOT requiring immediate notification (‘lA’) – Variations that do not require immediate notification may be submitted by the marketing authorisation holder (MAH) within 12 months after implementation.
Note:
a. Certain minor variations of Type IA require immediate notification after implementation, to ensure the continuous supervision of the medicinal product.
b. This should be submitted immediately and generally within 2 weeks of implementation of the change.
c. For quality changes, implementation is when the change was done in a quality system.
Examples of Type IAIN variation:
i. Change in the name and/or address of the marketing authorization holder
ii. Change in the name and/or address of a manufacturer/ importer of the finished product (including batch release or quality control testing sites)
iii. Changes in imprints, bossing, or other markings iv. Changes in the shape or dimensions of the pharmaceutical form; particularly immediate-release tablets, capsules, suppositories, and pessaries.
Examples of Type IA variation:
i. Addition of Physico-chemical test in the specification.
ii. Deletion of the non-significant test (ex: Identification test in Stability study).
iii. Tightening of specification limits (ex: Tightening of test limit for water content, residual solvents, and related substances, etc.
iv. Certificate of Suitability (CEP) updates/ renewal.
v. Finished Product (FP) batch size increase/ decrease within 10 fold for immediate-release products.
4.2 Type IB variations
Commission Regulation (EC) No 1234/2008 (‘the Variations Regulation’) defines a minor variation or Type 1B as a variation. This is neither a Type lA variation nor Type II variation nor an Extension.
Such minor variations must be notified to the National Competent Authority/ European Medicines Agency by the Marketing Authorisation Holder (MAH) before implementation.
Marketing Authorization Holder (MAH) must wait for 30 days to ensure that the notification is acceptable by the Agency before implementing the change (Tell, Wait and Do procedure).
Examples of Type IB variation:
i. Major change in the approved analytical method
ii. FP Mfg. site changes
iii. Shelf-life extension
iv. Change in storage condition
v. Minor changes to approved manufacturing process
vi. SmPC / PIL changes in-line with innovator product
4.3 Type II variations
Commission Regulation (EC) No 1234/2008 (‘the Variations Regulation’) defines a major variation of Type II as a variation that is not an extension and which may have a significant impact on the Quality, Safety, or Efficacy of a medicinal product.
Examples of Type II variation:
i. Addition of alternate/new API DMF supplier
ii. Relaxation of approved specification
iii. Major change in approved manufacturing process
iv. Major change in approved composition
4.4. Extension Applications:
Changes to a marketing authorisation listed in Annex I of Commission Regulation (EC) No 1234/2008 are regarded as “extensions” of the marketing authorisation.
Examples of Extension Applications:
i. Changes to the active substance(s):
Replacement of a chemical active substance by a different salt/ ester complex/ derivative with the same therapeutic moiety, where the efficacy/ safety characteristics are not significantly different.
ii. Change to strength, pharmaceutical form, route of administration:
a. Change of bioavailability;
b. Change of pharmacokinetics e.g. changes in rate of release;
c. Change or addition of a new strength/ potency;
d. Change or addition of a new pharmaceutical form;
e. Change or addition of a new route of administration.
f. Such applications will be evaluated in accordance with the same procedure as for the granting of the initial marketing authorisation to which it relates.
g. The extension can either be granted as a new marketing authorisation or will be included in the initial marketing authorisation to which it relates.
5. Timelines for approval of variation to marketing authorisation
Timelines for Type IA of variation approval
| Type IA | Timelines |
| Receipt of Type IA variation notification | Day 0 |
| Chicking starts | Day 1 |
| Acknowledgment of acceptance or refusal | by Day 30 |
Note: Type IA notification is a ‘Do and tell’ procedure. Hence, changes must be implemented prior to submission of notification
Timelines for Type IB of variation approval
| Type IB | Timelines |
| Receipt of Type IB variation notification | Day 0 |
| Chicking starts | Day 1 |
| The gernarl pre-assessment processing time is 14 days. Detailed assessment is done within 30 days. The outcome would be either approval or a request for further information (RFI) letter within the 30 days. | by Day 30 |
| Response to Request for Information (RFI) letter (If any) | Within 30 days |
| Assessment of the response submitted by firm | Within 30 days |
Note: This is a general timeline. This may differ based on circumstances.
Timelines for Type II of variation approval
| Type II | Timelines | Timelines | Timelines |
| Assessment Starts | Day 0 | Day 0 | Day 0 |
| Approval, refusal or withdrawal | Day 22 | Day 60 | Day 90 |
| Request for further information (RFI) (If any) | Day 21 | Day 59 | Day 89 |
| Timescale for receipt of response | Day 10 | Day 60 | Day 90 |
| Response received – clock on completion of processing | By 30 days | By 90 days | By 120 days |
Note: The timeline for Type II variations are 30-days, 60-days or 90-days depending upon complexity of the variation application.
6. References for types variations to the Marketing Authorisation in EU region
i. Variations Regulation (1234/2008) as amended by (712/2012)
ii. Variations Guidelines (2013/C 223/01)
iii. Guidelines of 16.05.2013 on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures.